Introduction
Small RNA seq (sRNA-seq) is a critical method for study of plant microRNAs and siRNAs. A typical experiment is analyzed by alignment to the relevant reference genome. As with most genomics experiments, qualitative visualization of the data is a critical part of the analysis. This is most readily accomplished with a genome browser. In this post, I will describe how “off the shelf” visualizations of plant sRNA-seq alignments fail to display the most important information. I will then describe several alternative methods to create more accurate and informative visualizations of genome-aligned sRNA-seq data. Links to the key tools are:
- The IGV genome browser
- My new sRNA_Viewer tools
- My script strucVis
The problems
Let’s assume you have a sRNA-seq analysis of two conditions, and thus have two different alignment files that you want to visually compare. The alignment files are in the standard BAM format, and they are both indexed. One very easy way to visualize these alignments is to use the Integrative Genome Viewer, which is a free download and very easy to set up on a laptop or desktop computer. Loading up two BAM files of sRNA-seq data with default settings on IGV will give you a view like this :

When given a BAM file, IGV will automatically calculate a coverage track, which is a barchart showing the depth of reads at each nucleotide position. IGV’s defaults also represent the strands of the individual alignments with different colors (blue and red). There are some problematic aspects to this display:
- Small RNA size cannot be rendered automatically by IGV. For plants, the size of the small RNAs is critical information: 21 nucleotide RNAs are strongly associated with post-transcriptional silencing, while 24 nucleotide RNAs are associated with transcriptional silencing. 22 nucleotide RNAs are associated with amplification of silencing by triggering secondary siRNA formation. And most sRNA-seq datasets contain large numbers of RNAs shorter than 20, or longer than 24 nucleotides; these off-sized RNAs are generally not involved in gene regulatory processes. To sum up, showing small RNA sizes is critical for interpretation of sRNA-sew alignments, but IGV cannot do this.
- The strand of the alignment is also critical. Plant siRNAs are processed out of longer double-stranded RNA precursors, and so they will align to both strands of the genome in a local cluster. In contrast, microRNAs are processed from stem-loop single-stranded RNA precursors, so they will align to a single genomic strand. Degraded bits of other RNAs, which are common in sRNA-seq datasets, will align to only one strand of the genome. The default IGV view does show strand of individual alignments (in color). However, this can be misleading when there is a large stack of alignments at the same position: IGV may be only able to render a small fraction of the reads (the rest are “stacked” out of the window), and the strands of the visible ones may not be a representative subset of the whole population. Ideally, we want the strandedness to be apparent in the coverage, not the individual alignments.
- Lack of space. It’s very difficult to show more than two BAM files on the same screen. But many experimental setups have many more than two alignments.
- Scaling. IGV will auto-scale each loaded BAM file independently. This makes it very hard to visually identify differences in small RNA accumulation when comparing BAM files.
Below, I’ll walk through several approaches that, collectively, address all of these issues and enable more informative visualization of plant sRNA-seq alignments.
Modifying BAM files by read length
The first step we take is to add a custom tag to each alignment in the BAM files. The tag “YS” was chosen by me to categorize each aligned small RNA. There are five categories defined:
- “<21nts” :: RNAs less than 21 nucleotides long. These are generally not known to be Argonaute-loaded and are thus more likely to be other types of RNAs, including degraded bits of random RNAs.
- “21nts” :: RNAs that are exactly 21 nucleotides long. In plants this size is associated primarily with post-transcriptional silencing without the triggering of secondary siRNAs.
- “22nts” :: RNAs that are exactly 22 nucleotides long. In plants this size is associated primarily with post-transcriptional silencing WITH triggering of secondary siRNAs.
- “23-24nts” :: RNAs that are exactly 23 or 24 nucleotides long. In plants these sizes are associated primarily with transcriptional silencing and RNA-directed DNA methylation that occurs in the nucleus.
- “>24nts” :: RNAs longer than 24 nucleotides. These are generally not known to be Argonaute-loaded and are thus more likely to be other types of RNAs, including degraded bits of random RNAs.
The script ‘add_YS.pl’ (available from the sRNA_Viewer GitHub repo) will add YS tags to a BAM file, like so: (more details are on the Github page)
add_YS.pl -b alignments.bam
Once modified with these custom tags, IGV visualizations can be improved. For instance, using the right-click menu for a BAM track in IGV, one can:
- ‘Group alignments by’ –> ‘tag’ and enter YS
- ‘Color alignments by’ –> ‘tag’ and enter YS
As shown below, this allows the aligned reads to be colored and grouped based on their size categories.

Grouping and coloring sRNA-seq reads by YS tag (size classes)
Switching the ‘Group alignments by’ to ‘Read strand’, both the read size categories and the strand can be visualized.
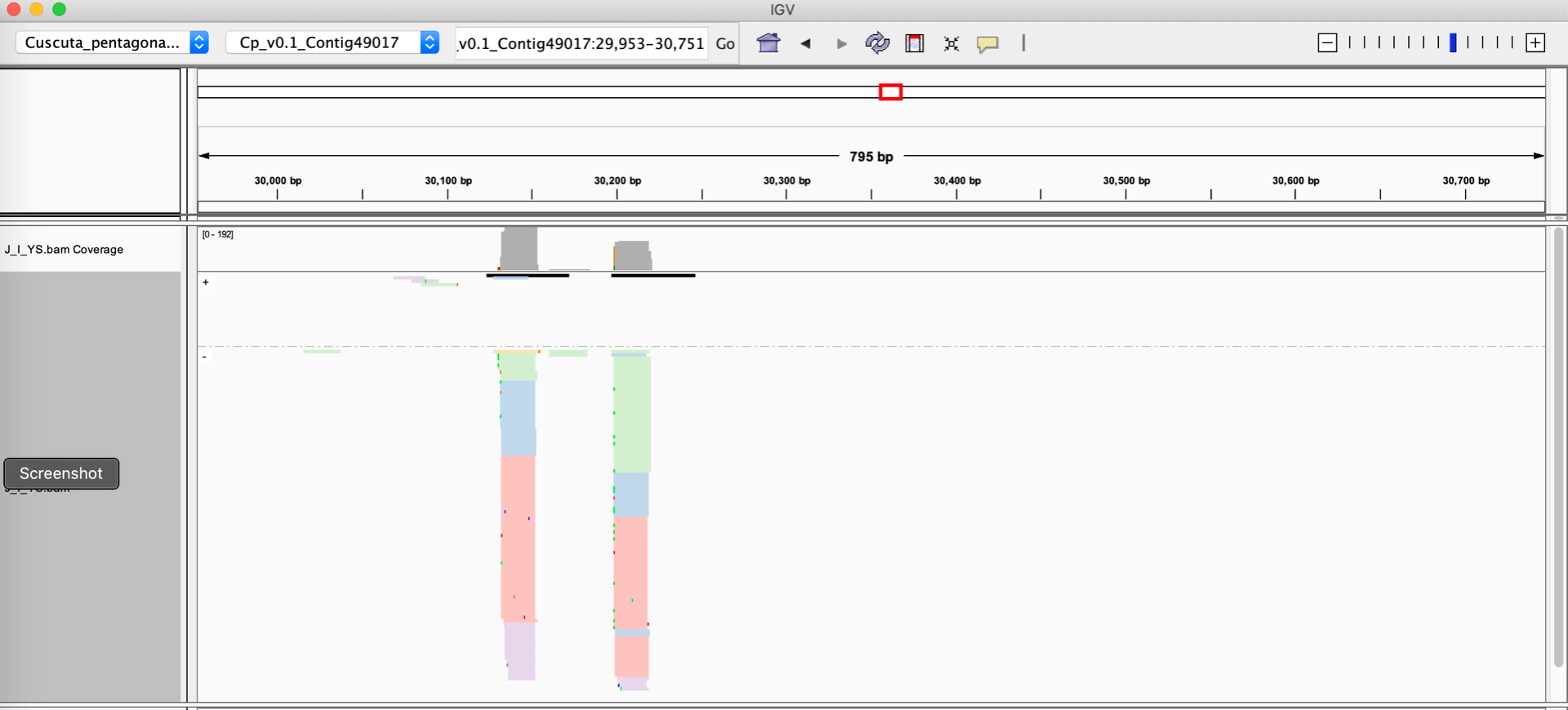
Coloring reads by YS tag (size category) and grouping them by Read strand.
Better views with sRNA_Viewer.R
Adding YS tags helps the IGV visualizations, but does not solve all of the problems. In particular, the problems of no room for more than ~2 tracks and different scalings between tracks remain. To solve this, I wrote a few more bits of code in the sRNA_Viewer repository. There is a two-step process that uses one or more YS-modified BAM files as input:
- Compute the per-nucleotide depth of coverage by strand, read-size category, and BAM file (script sRNA_coverage.pl) and output as a tab-separated text file.
- Use those data to make a coverage plot (script sRNA_Viewer.R).
The process can be run as a single pipeline (details given on the GitHub repo):
sRNA_coverage.pl -b bamfileList.txt -c Chr1:10993-11087 | sRNA_Viewer.R -p output.pdf
This will produce a PDF file where each BAM file has its own figure, the y-axis (RNA coverage in units of reads-per million) is scaled the same for all plots, and for which the strand is denoted by + and – numbers (for + strand and – strand reads, respectively). An example, with 12 different BAM files showing a microRNA locus that is differentially expressed in different libraries, is shown below:

Output from sRNA_Viewer.R , showing sRNA depth of coverage by size category and strand for 12 different BAM files.
Viewing sRNA alignment depth in the context of predicted precursor secondary structure
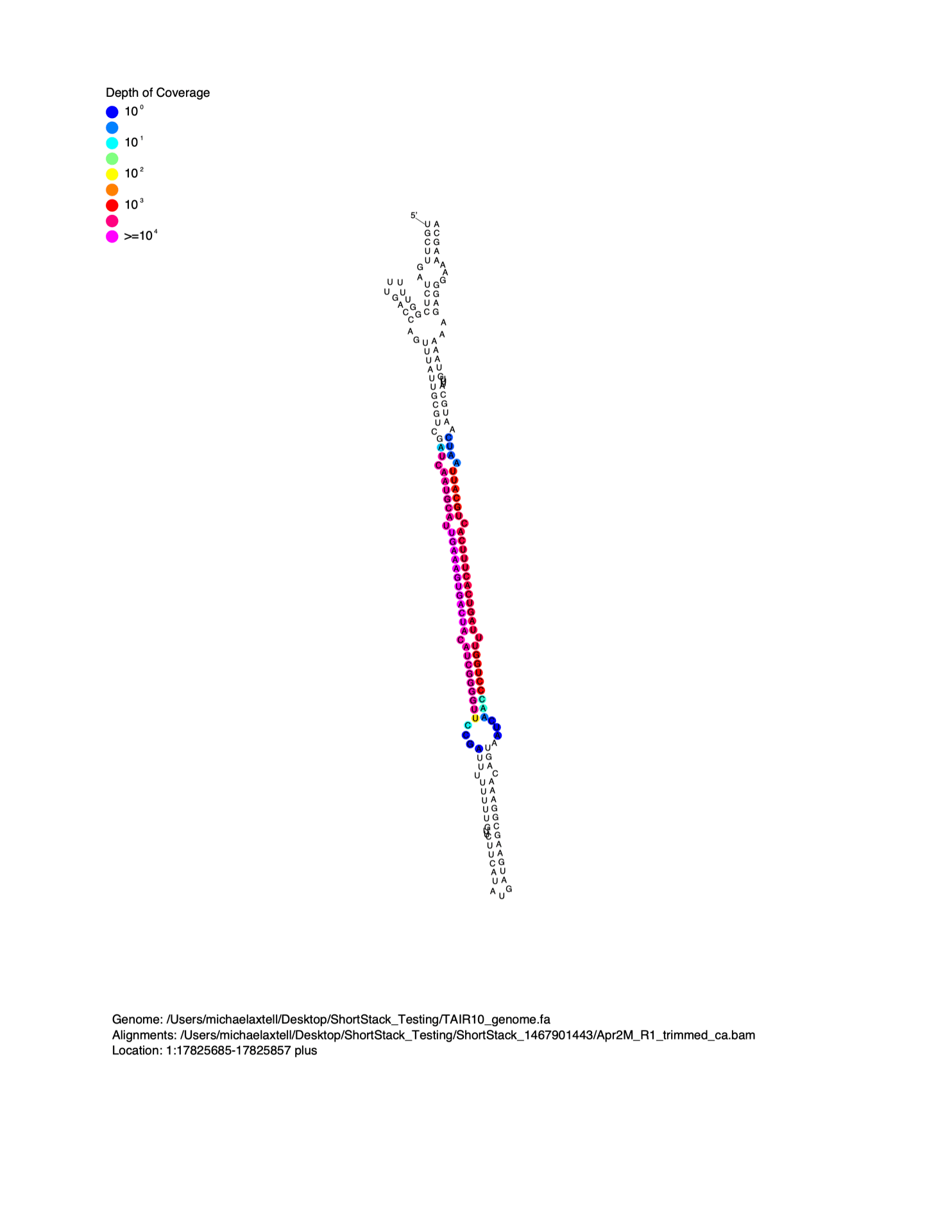
MicroRNAs are distinguished from short interfering RNAs (siRNAs) by the unique secondary structures of microRNA precursors. MicroRNAs are processed out of the helical regions of “stem-loop” RNA secondary structures in precursors. When viewed on a linear alignment (as in the examples above), this results in a characteristic “two-peak” pattern. The two peaks represent the microRNA, and the partner strand created during biogenesis (the so-called microRNA*). It is often helpful to visualize small RNA alignment depth as a function of predicted precursor RNA secondary structure. To do this, I wrote the script strucVis . This script takes in a BAM file, the corresponding genome (in FASTA format), coordinates and genomic strand of interest, and an optional name, and makes two outputs:
- A post-script formatted image showing the predicted RNA secondary structure of the locus, with sRNA-seq alignment depths color-coded on each nucleotide.
- A plain-text representation of the predicted RNA secondary structure with aligned sRNA reads shown underneath.
Details on the usage of strucVis can be found at the GitHub repository.
Example image from strucVis