The Facility’s PAGE equipment is free to use. If you don’t know how to run a gel, Tania can help!

Professor Ozbolat’s students, Kerim (left) and Madhuri (right) are using PAGE equipment with Donna’s (center) guidance.

The Facility’s PAGE equipment is free to use. If you don’t know how to run a gel, Tania can help!
Professor Ozbolat’s students, Kerim (left) and Madhuri (right) are using PAGE equipment with Donna’s (center) guidance.
Which reagent is better for reducing disulfides, DTT or TCEP? The two reagents are quite different in their reactivity, stability towards oxidation, reaction mechanisms, and other categories.

DTT is a thiol-containing reagent, and this must be considered in applications involving thiol labeling. TCEP is charged in solution and should not be used in isoelectric focusing. Aqueous solutions of TCEP are quite acidic (pH 2-3).
TCEP HCl is odorless, air-stable crystalline solid, soluble in water at a > 1 M concentrations. It reduces disulfides at room temperature in < 5 min in dilute solutions (5 -50 mM). There is no need to remove TCEP prior to the use of sulfhydryl-reactive labels or crosslinkers. TCEP is selective toward disulfides, and is reactive at a broad pH range.
For those who love Chemistry:

Reduction of disulfide with TCEP. First step is rate-determining, kinetic rather than thermodynamic control.

Reduction of disulfide with DTT. Formation of stable cyclic disulfide drives the reaction.
You can learn more about DTT and TCEP from this article: “A Comparison between the Sulfhydryl Reductants Tris(2-carboxyethyl)phosphine and Dithiothreitol for Use in Protein Biochemistry”, Elise Burmeister Getz et al. Analytical Biochemistry 273, p. 73–80 (1999)
Immobilized pH gradient (IPG) strip rehydration: active time 5 min per sample, rehydration is carried out overnight
Isoelectric focusing (IEF): active time 5 min per sample, IEF 5-6 hrs.
Preparation for the 2nd dimension SDS PAGE: 1 hr.; necessary reagents and solutions are prepared during the last hour of IEF.
SDS PAGE, staining, and de-staining: 2.5 hrs. with de-staining overnight.
Protein sample should be lyophilized or precipitated. To keep the ionic strength of the protein solution at minimum, avoid salts. Non-ionic and zwitterionic solubilizing agents could be present, but keep in mind solubility limits.
Protein load for a complex sample to be stained with Coomassie is approximately 100 ug. The protein load varies depending on many factors including stain sensitivity, IPG range, or downstream applications (refer to the manual).
Protein sample is dissolved in the sample buffer (Bio-Rad cat #163-2106), 125 ul for 7 cm IPG strips and 185 uL for 11 cm IPG strips. The protein solution is pipetted into a channel in a rehydration tray. An IPG strip is placed gel side down into the channel, covered with mineral oil, and left overnight at 4 C.
IPG strips are placed into a focusing tray, covered with mineral oil, and the tray is placed into the IEF cell for 5-6 hrs.
After the IEF is complete, strips are soaked in a reducing buffer, followed by alkylating buffer, rinsed in SDS running buffer, and placed at the top of an SDS-PAGE gel. A molten agarose solution is applied to the well. Once agarose solidifies, the gel is ready for the 2nd dimension electrophoresis.
After completion of the SDS-PAGE, gels are rinsed, stained, and destained. Alternatively, the gels can be electro-blotted.
What IPG strip is the best for your experiment? Start by choosing the length (7 cm or 11 cm). Short strips are compatible with the Mini-PROTEAN cell, longer strips are compatible with the Criterion cell. All parameters being equal, a larger gel affords better resolution. Next select pH range depending on a type of your experiment: broad range (e.g. 3-10) for a global view, narrow range for a zoom-in view.
Bio-Rad cat #163-2000, 7 cm, pH 3–10, immobilized pH gradient (IPG) strip for first-dimension separations, pkg of 12
Bio-Rad cat #163-2014, 11 cm, pH 3–10, immobilized pH gradient (IPG) strip for first-dimension separations, pkg of 12
Rehydration/sample buffer, Bio-Rad cat #163-2106
Agarose, Bio-Rad cat #163-2111
Criterion Tris-HCl Gel, Bio-Rad cat #345-0040, Pkg of 1, 8–16% polyacrylamide gel, prep+2 well, 800 μl, 13.3 x 8.7 cm (W x L), for use with Criterion and Criterion Dodeca cells
10x Tris/Glycine/SDS, Bio-Rad cat #161-0732, 1 L, 10x premixed electrophoresis buffer, contains 25 mM Tris, 192 mM glycine, 0.1% SDS, pH 8.3 following dilution to 1x with water
SDS equilibration buffer II: 6M urea, 2% SDS, 0.375 M Tris-HCl (pH 8.8), 20% glycerol, Bio-Rad cat #163-2108
And for the best results: Electrode Wicks, Bio-Rad cat #165-4071, Pkg of 500, precut electrode wicks, for use with the PROTEAN® i12™ IEF system. (I still have about 450, no need to buy)
2D PAGE detailed procedure for the Bio-Rad starter kit
Please let me know if you are interested in a 2D PAGE experiment!
As promised, here’s a straightforward way to remove detergents, urea, and other LC-MS incompatible nasties from small-volume samples. The literature calls it ‘gel-assisted’ proteolysis. The idea is to entrap the protein solution in a polyacrylamide gel matrix, wash out detergents, salts, and chaotropic agents, and perform in-gel digestion. This technique works great for membrane proteins which are notoriously difficult to dissolve, and it is quite useful for any protein sample clean-up.

For my little demo, I used a 1 mg/mL BSA solution in 2% SDS. The disulfides were reduced with TCEP and alkylated with IAA, after which the protein solution was very thoroughly mixed with a 30% T acrylamide monomer solution. I quickly added 10% APS and TEMED and immediately vortexed and centrifuged this mixture so that the liquid is collected at the bottom of the tube. The polymerization time is very short, a minute or two! I left it to completely polymerize for another 20 min.

Using scalpel I removed the gel plug from the tube and diced it into small pieces. After 6 washes with 8 mg/mL ammonium bicarbonate in 50% acetonitrile, I dried the gel pieces in neat acetonitrile, removed the acetonitrile and added trypsin (see the in-gel digestion tutorial for details).
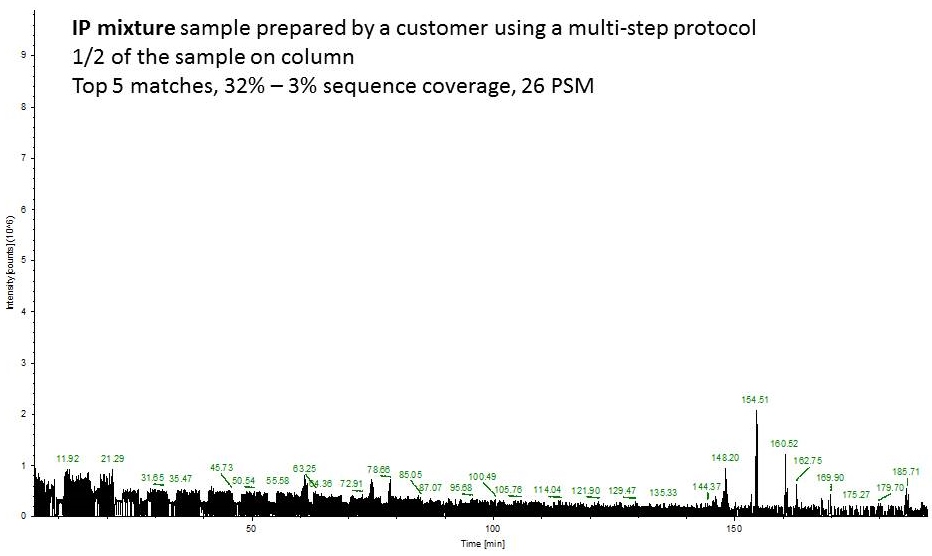
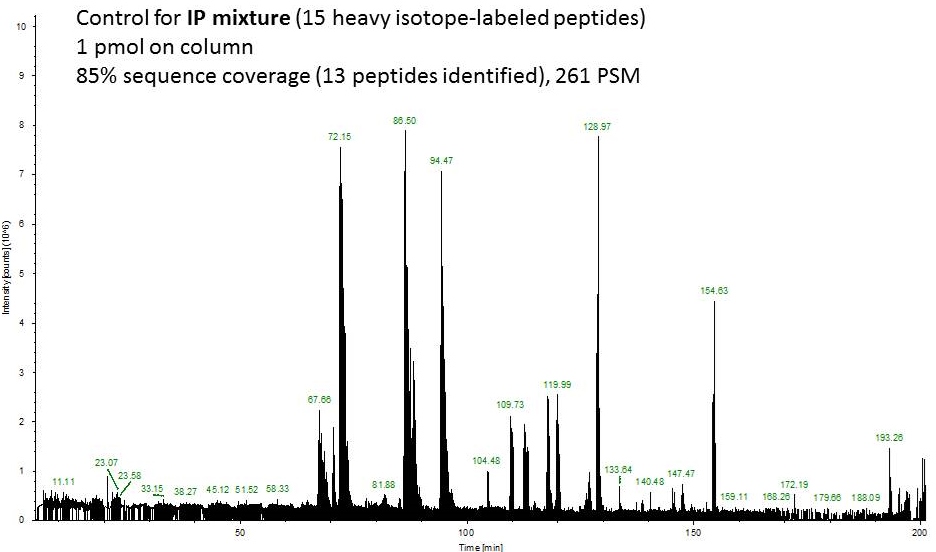
Fast forward to the MS analysis: Since the original BSA solution was very concentrated, I dissolved the peptides in 540 uL of mobile phase and injected 1 uL of this solution (55 ng total protein on column or approximately 1 pmol).

Second example shows an ion chromatogram from a 10-uL IP eluent containing 2% detergent which I cleaned up and digested using this technique.

As always, let me know if you have questions!
References:
doi: 10.1074/mcp.M500138-MCP200
doi: 10.1074/mcp.M800068-MCP200
First, let’s define ‘large’. The Proteome Discoverer sets the upper limit for a precursor ion at 10,000 Da. This means anything bigger than 10 kDa will not be considered even if it’s present in the MS data, and a different software package ($$) will be required to analyze the high MW data. Clearly, proteolytic peptides with MW > 10 kDa will not be very useful for protein identification. I suggest using a different enzyme or a combination of enzymes. I have seen tryptic peptides up to 7 kDa in some in-gel digested samples, so apparently some large peptides do come out of gel.
Next, a large peptide’s physico-chemical properties (e.g. hydrophobicity, pI, hydrodynamic radius) must be considered as they will affect the extraction efficiency. If the peptide’s properties are known, the extraction solvent composition and pH can be adjusted to improve the peptide’s solubility.
Finally, let’s consider the gel from which the large peptides need to come out. Obviously, it will be easier to get the large peptides out of a 4 %T gel than out of a 20 %T one. Soaking a gel piece in deionized water and then freezing it should crash enough pores in the gel to improve the extraction of large peptides (water expands as it freezes). Additionally, the gel could be ‘squeezed out’ a few times by changing extraction solvent from neat acetonitrile to an aqueous mixture. The gel piece will shrink in acetonitrile expelling the peptide solution. Re-hydrating the gel and then shrinking it again in acetonitrile will ‘squeeze out’ more of the digest.
Using elevated temperature (50 C), vortex mixer, and/or ultrasonic bath should all improve the extraction. Use common sense: 50C and a high pH buffer is not a good idea for the phosphopeptide extraction. Another word of caution: don’t get carried away. Three extraction steps should be enough. If you end up with a large volume (e.g. more than 0.5 mL), the benefits of a thorough extraction might become negated by the losses due to dilution. Peptides and proteins tend to adhere to the polypropylene tubes. A large volume of a dilute peptide solution presents a large surface area for the peptides to adsorb.
What to do if this doesn’t work? You can try in-solution digestion. If the mixture is too complex and a PAGE step is necessary, you can try electroeluting the protein(s). Intact proteins electroeluted from gel bands can be buffer-exchanged using small-volume 3,000 Da MWCO spin columns and proteolyzed in solution.
If you have never done in-gel protein digestion, this tutorial is for you! The protocol is really simple and does not require any specialized equipment. Even if your gel has been sitting in the fridge for a month or two, it should still work, but no mold, please! First order of business is to choose a band and cut it out. You will need a clean blade, a clean surface (a clean glass plate or a clean transparency sheet), a clean microcentrifuge tube, and a pair of clean gloves. Everything should be clean: the goal is to minimize contamination of interesting proteins by uninteresting keratins.
1. Place the gel on a glass plate, blot excess water with a clean paper tissue. Select a band and cut out only the stained portion of the gel. Avoid unstained area: it will sponge up a protease solution, giving nothing interesting in return.
2. Dice the band into 1-mm (1/16”) sections; this will help the protease reach more protein inside the gel.
3. Place the gel cubes in a clean microcentrifuge tube, cover with water to prevent them from drying out, and label the tube with no more than 5 characters. For example, use your initials and numbers.
4. Include a positive control! There is no charge for analyzing a control! Cut out a known protein band following steps 1, 2, and 3.

The in-gel digestion protocol can be found on Thermo website along with the product numbers for all necessary reagents.
You will need 50% acetonitrile (ACN) solution containing 8 mg/mL ammonium bicarbonate (wash solution). Add 0.05 – 0.1 mL of this solution per gel band, enough to completely cover the gel. Incubate at 37 C for 15-20 min, discard the solution. Repeat two more times. At this point, all or most of the blue color should be gone. If you are destaining SYPRO, there is no easy way to tell whether it is all gone, of course.
You will need
Cover the gel slices with 5 mM TCEP and incubate 10 min at 60 C. Remove the TCEP solution and cover the gel with 100 mM IAA solution. Incubate at 37 C for 15 min with occasional shaking, preferably protected from light. Room temperature will also work, but give it extra 10-15 min. Discard the IAA solution and wash the gel with the wash solution three times to remove IAA and TCEP. (In the Pierce protocol, you might notice that 100 mM IAA requires 9.3 mg in 1 mL, while MW of IAA is 184.9. It is a typo. Either way: 50 mM, 9.3 mg/mL or 100 mM, 18 mg/mL, will work as there will be a large molar excess of IAA compared to Cys. Shrink the gel by covering it with ACN and incubating at r.t. for 15 min or until the gel turns white and brittle. Remove ACN and allow the gel to air dry for 15 min at 37 C (or longer at room temperature).
Make 1 mg/mL trypsin stock solution in 50 mM acetic acid or 1 mM hydrochloric acid. This solution can be aliquoted and stored in a freezer for months. Dilute trypsin to 0.01 mg/ml (1:100 dilution) with 8 mg/mL ammonium bicarbonate. Add 50 uL of this solution per gel band and incubate at 37 C 8-24 hours. If you are working with a large piece of gel or several bands combined in one tube, make sure that after the gel re-hydrates in the enzyme solution, it is completely covered. Add more enzyme solution if necessary.
You will need 50% ACN solution containing 0.1% formic acid (FA). If you don’t have formic acid in your lab, let me know – we will share ours with you. Transfer the digest solution to a new tube. Extract each gel band with 50 uL of 50% ACN/0.1% FA (more, if working with a large volume of gel) by incubating at 37 C for 15 min. Transfer this solution to the new tube with the digest solution. Repeat 2 more times. Evaporate the combined extracts in a vacuum concentrator (e.g. Speedvac). Please note, if you are extracting large peptides (>5000 Da), using a sonic bath may be a better option than incubation. Also, in addition to the three 50% ACN extractions, you can use 100% acetonitrile for the final extraction. Submit your dried samples along with a picture of the gel so that I can estimate how much of each sample to use for analysis, and don’t forget to fill out the Protein ID Request form
As always, let me know if you have any questions!

I get a lot of inquiries about our PAGE (polyacrylamide gel electrophoresis) equipment, so here are some information on the equipment we have and the supplies we use.
We share our PAGE equipment with you at no charge. You need to purchase your own supplies: e.g. gels, standards, buffers, and other consumables. Sometimes we have supplies left over from the Facility experiments, which we will gladly share with you as well. Please, ask before you buy your own!
Our power supply is a basic model; it does not store programmed methods. It can run up to 4 cells simultaneously, as long as the conditions are the same.
We have three Bio-Rad electrophoresis cells, a Mini-PROTEAN Tetra for up to 4 mini-gels (8.6 x 6.7 cm), a Criterion cell for up to 2 midi-gels (13.3 x 8.7 cm), and a Criterion Dodeca cell for up to 12 midi-gels. We also have a blotter for 13.3 x 8.7 cm gels.
Our PROTEAN IEF cell (Bio-Rad) is capable of storing methods, and accepts 7 cm, 11 cm, and 18 cm focusing trays. We also have rehydration trays for all three IPG (immobilized pH gradient) strip lengths.
You can use our gel casting stands and glass plates (0.75 mm) to cast your own mini-gels. In my experience, 1-mm thick gels hold up much better, especially for low %T (4-10%). Bring your own plates if you wish!
The Pierce precast mini gels from Fisher are compatible with the Bio-Rad Mini-PROTEAN cell. We do not buy precast gels from Bio-Rad due to the high shipping costs. Fisher orders ship free. You can learn more here.
Immobilized pH gradient (IPG) strips are also available from Fisher (GE Healthcare Immobiline DryStrip gels, for example). The prices are comparable with the Bio-Rad’s, and the shipping is free.
05/28/2014 ETA: According to the GE Healthcare website, Immobiline IPG strips and buffers are not compatible with the Bio-Rad IEF cell running conditions. I am not familiar with the GE system and cannot tell you whether this is true; so to be safe, please use Bio-Rad IPG strips and buffers. If anyone has successfully run Immobiline strips on Bio-Rad IEF cell, please share with us! This information could save your colleagues both time and money!
Disclaimer: We do not receive any form of compensation for promoting any brand or a particular distributor.
Still have questions? Let’s meet and talk about your project!