
If you are interested in learning how proteins are identified using mass-spectrometry-based proteomics, Proteome Software website has an excellent collection of short and informative essays.
Tag Archives: protein identification
FAQ: Why should I dice gel bands?

In-solution digestion of proteins
Purified proteins or protein mixtures can be digested in solution if an additional separation step is undesirable or unnecessary.
Proteins in solution are usually denatured by boiling or using denaturing buffers. During this step, the disulfide bonds must be reduced, and the sulfhydryl groups must be alkylated to prevent the disulfides from re-forming. The protein samples are then incubated with trypsin for several hours, and the resulting peptides can be analyzed by MS.
Denaturing buffers contain chaotropic agents, salts, and detergents at concentrations that inactivate trypsin. Before adding trypsin, you should desalt your protein sample and remove detergents. There are a number of the detergent removal and desalting options: detergent removal spin columns, size-exclusion and MW-cut-off spin columns, ion-exchange membranes and resins, etc. Gel-assisted proteolysis is another option, but then it is not really an ‘in-solution’ procedure, although it does not involve electrophoresis. I thought I should mention it here in case it could be of interest to you.
You will need
Digestion buffer: 16 mg/mL ammonium bicarbonate in water
Reducing reagent: 30 mg/mL TCEP (~100 mM, Sigma C4706) or 15 mg/mL DTT (Sigma D0632 ) in digestion buffer. NOTE: The 30 mg/mL TCEP stock solution must be prepared in 16 mg/mL (~200 mM) ammonium bicarbonate to bring up its pH. The final concentration of TCEP in the digestion mixture should be 5-10 mM.
Alkylating reagent: 18 mg/mL iodoacetamide (Sigma I1149) prepared fresh in the digestion buffer
Proteomics grade trypsin (e.g. Sigma T6567-5x20UG or Thermo Pierce 90057, 5 vials x 20 ug lyophilized powder). Trypsin, 20 ug can be dissolved in 20 uL of 1 mM HCl or 50 mM acetic acid, pH ~ 3, aliquoted and stored at -20C (stock solution).
To prepare activated (or working) trypsin solution, dilute trypsin stock solution with digestion buffer 10-fold to 0.1 ug/uL concentration.
Procedure
Volumes are approximate, it is a sample procedure after all. Trypsin should not exceed 5% of the total protein, provided the protein concentration range is known.
Combine 15 uL digestion buffer, 3 uL reducing reagent, and up to 12 uL sample solution containing 0.025 – 10 ug protein (total volume 30 uL)
Denature/reduce at 50-60 C (TCEP) or in a boiling water bath (DTT) for 5 – 10 min, cool to r.t., spin down to collect the sample
Add 3 uL alkylating reagent and incubate in the dark at r.t. for 20 min
If protein sample contains detergents, salts, or chaotropic agents, perform buffer exchange after the alkylation using a 3,000 MWCO centrifugal filter. It will be impossible to remove detergents after the digestion; and most detergents are not compatible with LC MS analysis. You can find a list of MS-compatible detergents here.
Add 1-5 uL activated trypsin and incubate at 37 C for 3 hrs. Optional: add 1-5 uL of fresh activated trypsin and incubate for an additional 2 hrs at 37 C or overnight.
Once the incubation is complete, the peptides can be submitted for analysis or stored at -20 C.
Helpful tips
Minimum sample amount required for MS analysis is in the fmol/uL range (ng/uL). Solutions of peptides at very low concentrations (e.g. less than 100 fmol/uL) should not be stored for more than 1-2 days.
Always run a control along with your sample. It could be a 1 mg/mL solution of bovine serum albumin or other standard protein that you have in your lab prepared in the same buffer as your sample and taken through the entire procedure. We don’t charge for analyzing your controls.
TCEP is a great reducing reagent because it does not contain -SH groups and thus does not consume iodoacetamide during alkylation, unlike DTT. TCEP solutions in water are acidic. Depending on your buffer composition, you might observe your sample coming out of the sample tube as a soapy foam the moment you add TCEP. It is pretty much impossible to put that foam back into the tube, don’t ask me how I know. So, prepare your TCEP solutions in ammonium bicarbonate buffer to get a pH close to 8. Another note about TCEP: it should never be stored in phosphate buffers because it quickly decomposes in the presence of phosphate.
Have I missed anything? Let me know!
Proteomics sample volume and concentration
Every sample is different in terms of purity and structural and compositional complexity. The MS detection sensitivity of a routine analysis is in a range of 0.1 to 10 pmol of protein. Some peptides ionize and/or fragment more efficiently and will produce good spectra at 0.1 pmol per injection while other peptides may be completely ‘invisible’ even at 100 pmol per injection. Keep in mind that it is always easier and faster to dilute a sample than to concentrate it.
For routine analyses, a 1-microliter injection is usually made unless the protein concentration is known and requires a larger-volume injection. The injection volume cannot exceed 6 microliters.
For simple mixtures (in-solution digested purified or enriched proteins and in-gel digested protein bands), 3 to 10 microliters of sample must be submitted because smaller volumes tend to dry out. This is only true of expertly prepared samples that do not require purification and/or filtration.
If you are planning to analyze a complex mixture, please contact Tatiana.

Protein ID report
The very first time you receive your Excel file(s) summarizing the search results, you might feel confused. No worries, based on my experience, you are not alone! This post will guide you through.
My file naming system is as follows: date-sample name-number in the queue. For example, a positive control for your sample analyzed on January 29, 2014 would be named “14-01-29-CTRL-04”
You should receive twice as many files as the number of samples you submitted. Your sample is always injected after a blank run. This is done to account for potential carryover from previous sample injections, which is unavoidable in a service facility environment. Thus, “14-01-29-blank-01” precedes your first sample, “14-01-29-TNL1-02” and so on. I may also include my standard data so that you could see what type of data is obtained using a pure standard. Blanks, samples, your controls, and my standards are always run using the same instrument parameters.
Proteins page
When you open your Excel file, you should see a list of proteins each of which has the following parameters:
Accession
UniprotKB protein accession number, the unique identifier assigned to the protein by the FASTA database used to generate the report. To find the NCBInr equivalent, copy the accession number and paste it into NCBInr search, selecting ‘protein’ from the drop-down list
Description
UniprotKB protein description. Provides the name of the protein exclusive of the identifier that appears in the Accession column.
Score
The protein score, which is the sum of the scores of the individual peptides. I use SEQUEST search algorithm, for which the score is the sum of all peptide Xcorr values above the specified score threshold. The score threshold is calculated as follows: 0.8 + peptide_charge × peptide_relevance_factor where peptide_relevance_factor is a parameter with a default value of 0.4. For each spectrum and sequence, the Proteome Discoverer application uses only the highest scored peptide. When it performs a search using dynamic modifications, one spectrum might have multiple matches because of permutations of the modification site. (The higher the better)
Coverage
The percent coverage calculated by dividing the number of amino acids in all found peptides by the total number of amino acids in the entire protein sequence. (The higher the better)
# Proteins
The number of identified proteins in the protein group of a master protein. Proteins are grouped based on sequence homology and/or isoforms as explained below.
# Unique peptides
The number of peptide sequences unique to a protein group.
# Peptides
The number of distinct peptide sequences in the protein group.
# PSMs
The total number of identified peptide sequences (peptide spectrum matches) for the protein, including those redundantly identified. (The higher the better)
# AAs, MW [kDa], calc. pI
The calculated parameters of the protein based on the amino acid sequence in the FASTA database used to generate the report. The Proteome Discoverer application calculates the molecular weight without considering post-translational modifications. If you have separated proteins by molecular weight by PAGE, you can use the protein’s molecular weight as a rough constraint to estimate whether it is reasonable to identify a particular protein in a certain fraction that was analyzed.
Peptides page
Next, expand the sheet by clicking on [+] which opens the column parameters for the associated peptides.
A2 or other Letter/number, first column
A top level confidence achieved with the peptide sequence: high confidence, medium confidence, or low confidence. I send you only the high-confidence data, unless instructed otherwise.
Sequence
The sequence of amino acids that compose the peptide.
# PSMs
The total number of identified peptide sequences (PSMs) for the protein, including those redundantly identified. (The higher the better)
# Proteins
Displays the number of proteins in which this peptide is found
# Protein Groups
The number of protein groups in which this peptide is found.
MS/MS-based proteomics studies are based on peptides. However, deducing protein identities from a set of identified peptides could be difficult because of sequence redundancy, such as the presence of proteins that have shared peptides. These redundant proteins are automatically grouped and are not initially displayed in the search results report.
The proteins within a group are ranked according to the number of peptide sequences, the number of PSMs, their protein scores, and the sequence coverage. The top-ranking protein of a group becomes the master protein of that group. By default, only the master proteins are displayed on the Proteins page.
A protein group consists of the following:
- One master protein that is identified by a set of peptides that are not included (all together) in any other protein group.
- All proteins that are identified by the same set or a subset of those peptides.
The # Proteins column on the Proteins and Peptides pages of the results report displays the number of identified proteins in the protein group of a master protein.
Protein Group Accessions
The unique identifiers (accessions) of all master proteins from all protein groups that include this peptide sequence. Since I normally group proteins by selecting the “Consider Leucine and Isoleucine as Equal” option, this column also lists identifiers from master proteins that may include this specific peptide sequence. The identifiers displayed in the Protein Group Accessions column are the same as those displayed in the Accession column on the Proteins page.
Modifications
The static and dynamic modifications identified in the peptide. I always use iodoacetamide for Cys alkylation, and this static modification will be in your search results as ‘Carbamidomethyl’ unless you modified your Cys residues with a different reagent. Met oxidation and Asn and Gln deamidation are common dynamic modifications.
delta Cn
The normalized score difference between the currently selected PSM and the highest-scoring PSM for that spectrum. (The lower the better)
Xcorr
A search-dependent score. It scores the number of fragment ions that are common to two different peptides with the same precursor mass and calculates the cross-correlation score for all candidate peptides queried from the database by SEQUEST searches. (Jimmy K. Eng, Ashley L. McCormack, and John R. Yates, III; An Approach to Correlate Tandem Mass Spectral Data of Peptides with Amino Acid Sequences in a Protein Database. J. Am. Soc. Mass Spectrom. 1994, 5, 976-989) (The higher the better)
Probability (not available in our version)
The probability score for the peptide. This score is an assessment of the probability that the reported match is a random occurrence. A lower probability score indicates a better match. (The lower the better)
Charge
The charge state of the peptide, z (z is always greater than 1 as set during the MS analysis).
MH+ [Da]
Calculated m/z of the peptide with z = 1. It should be “MH+ [m/z]”, not [Da].
delta M [ppm]
Mass measurement error in parts per million, ppm (The lower the better)
RT [min]
The peptide’s retention time during chromatographic separation.
# Missed Cleavages
The number of cleavage sites in a peptide sequence that a cleavage reagent (enzyme) did not cleave. This number excludes cases where an amino acid (e.g. Pro) inhibits the cleaving enzyme (e.g. trypsin).
If you prefer the original user guide, I have included it for your reading pleasure.
Extracting large peptides from gel after digestion
First, let’s define ‘large’. The Proteome Discoverer sets the upper limit for a precursor ion at 10,000 Da. This means anything bigger than 10 kDa will not be considered even if it’s present in the MS data, and a different software package ($$) will be required to analyze the high MW data. Clearly, proteolytic peptides with MW > 10 kDa will not be very useful for protein identification. I suggest using a different enzyme or a combination of enzymes. I have seen tryptic peptides up to 7 kDa in some in-gel digested samples, so apparently some large peptides do come out of gel.
Next, a large peptide’s physico-chemical properties (e.g. hydrophobicity, pI, hydrodynamic radius) must be considered as they will affect the extraction efficiency. If the peptide’s properties are known, the extraction solvent composition and pH can be adjusted to improve the peptide’s solubility.
Finally, let’s consider the gel from which the large peptides need to come out. Obviously, it will be easier to get the large peptides out of a 4 %T gel than out of a 20 %T one. Soaking a gel piece in deionized water and then freezing it should crash enough pores in the gel to improve the extraction of large peptides (water expands as it freezes). Additionally, the gel could be ‘squeezed out’ a few times by changing extraction solvent from neat acetonitrile to an aqueous mixture. The gel piece will shrink in acetonitrile expelling the peptide solution. Re-hydrating the gel and then shrinking it again in acetonitrile will ‘squeeze out’ more of the digest.
Using elevated temperature (50 C), vortex mixer, and/or ultrasonic bath should all improve the extraction. Use common sense: 50C and a high pH buffer is not a good idea for the phosphopeptide extraction. Another word of caution: don’t get carried away. Three extraction steps should be enough. If you end up with a large volume (e.g. more than 0.5 mL), the benefits of a thorough extraction might become negated by the losses due to dilution. Peptides and proteins tend to adhere to the polypropylene tubes. A large volume of a dilute peptide solution presents a large surface area for the peptides to adsorb.
What to do if this doesn’t work? You can try in-solution digestion. If the mixture is too complex and a PAGE step is necessary, you can try electroeluting the protein(s). Intact proteins electroeluted from gel bands can be buffer-exchanged using small-volume 3,000 Da MWCO spin columns and proteolyzed in solution.
Gel bands and gel bandits
A protein ID confirmation is probably the most requested proteomics service in the facility; and it is not just a good (or ‘expensive’) idea – it could save you hours in the lab in the long run. When the MS result is not what you expected, don’t panic: use the MS information to your advantage! Knowing the parameters of interfering proteins (MW, pI, mechanism of metal ion binding) can help you to optimize or change your purification scheme.
…Poly-His purification approach was inspired by high affinity of transition metal ions (divalent Co, Ni, Zn, and Cu) for His and Cys residues in naturally occurring proteins way back in 1975 …
What I usually see is a prominent gel band that is thought to contain a protein of interest with a poly-His purification tag which has been expressed in E.Coli and purified on an immobilized metal affinity chromatography (IMAC) column. When the protein of interest ID is confirmed, you leave me with a smile and a thank you, so read no further.
This post is for my disappointed customer whose gel band got hijacked by a bunch of E. Coli gel bandits. Having seen enough of their sneering mug shots/accession numbers, I have compiled a quick reference list of these interfering so-and-so’s from the references 1 and 2 (also included as pdf). Please note that these accession numbers are for K12, other strains will have a different accession number for the same gene product. Additional references, some of which offer solutions to the interference problem(s), are included for your enjoyment. If you encounter recurring interfering proteins in your purification system, and they are not listed here, please share this information!
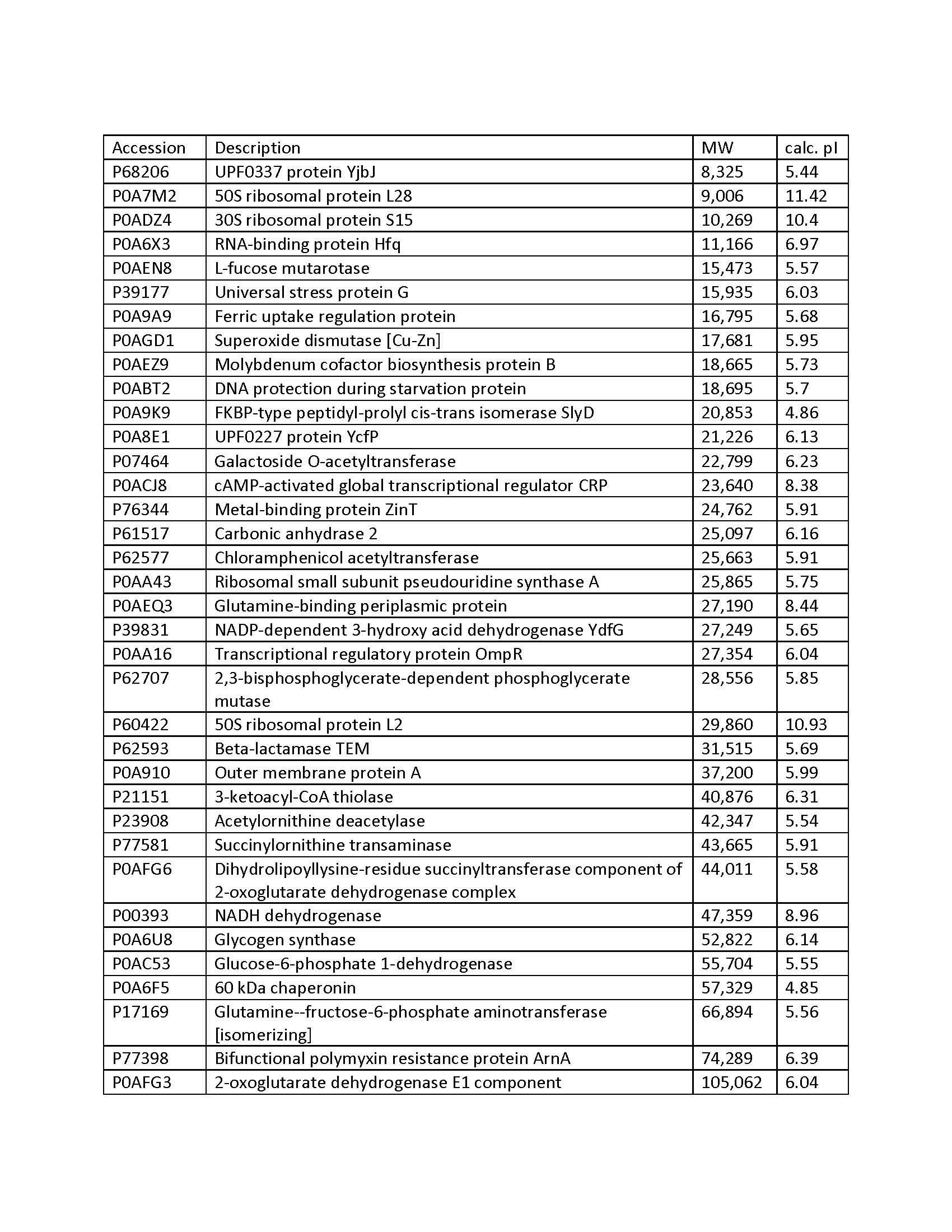
References
- Bolanos-Garcia and Davies; Structural analysis and classification of native proteins from E. coli commonly co-purified by immobilised metal affinity chromatography. doi:10.1016/j.bbagen.2006.03.027
- Bartlow et al.; Identification of native Escherichia coli BL21 (DE3) proteins that bind to immobilized metal affinity chromatography under high imidazole conditions and use of 2D-DIGE to evaluate contamination pools with respect to recombinant protein expression level. doi:10.1016/j.pep.2011.04.021
- Robichon et al.;Engineering Escherichia coli BL21(DE3) derivative strains to minimize E.coli protein contamination after purification by immobilized metal affinity chromatography. doi:10.1128/AEM.00119-11
- Parsy et al.; Two-step method to isolate target recombinant protein from co-purified bacterial contaminant SlyD after immobilised metal affinity chromatography. doi:10.1016/j.jchromb.2007.03.046
- Block et al.; Immobilized-Metal Affinity Chromatography (IMAC): A Review. Methods in Enzymology, doi:10.1016/S0076-6879(09)63027-5
Gel bandits reference list pdf
Highlights from today’s seminar
Direct analysis of TLC spots by MALDI-TOF



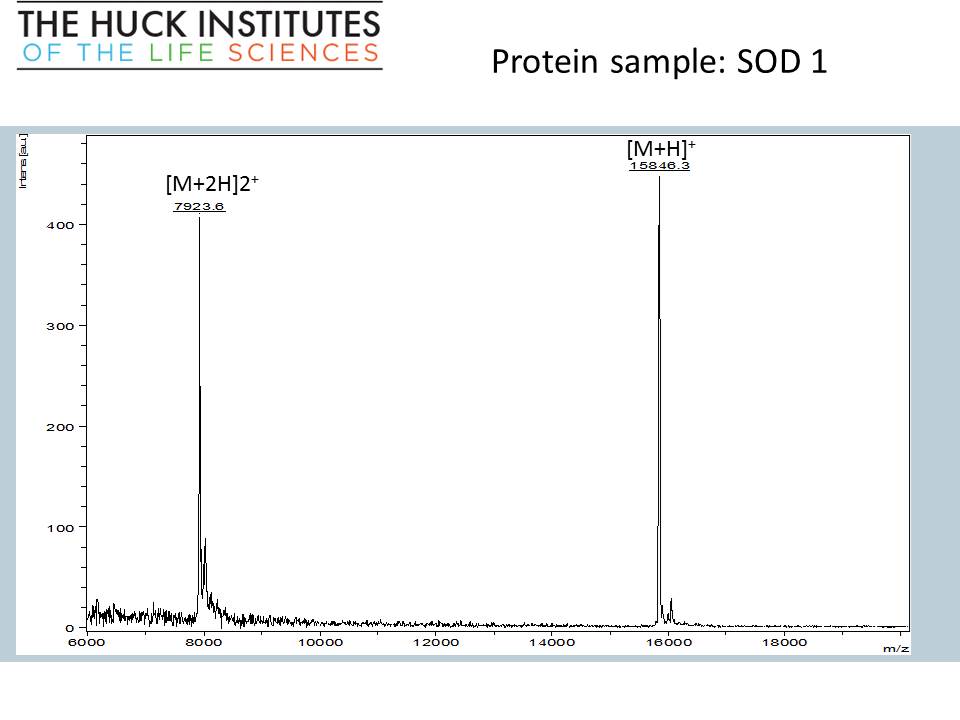
“Edmass” sequencing, N-terminal and C-terminal sequencing of intact SOD1 protein


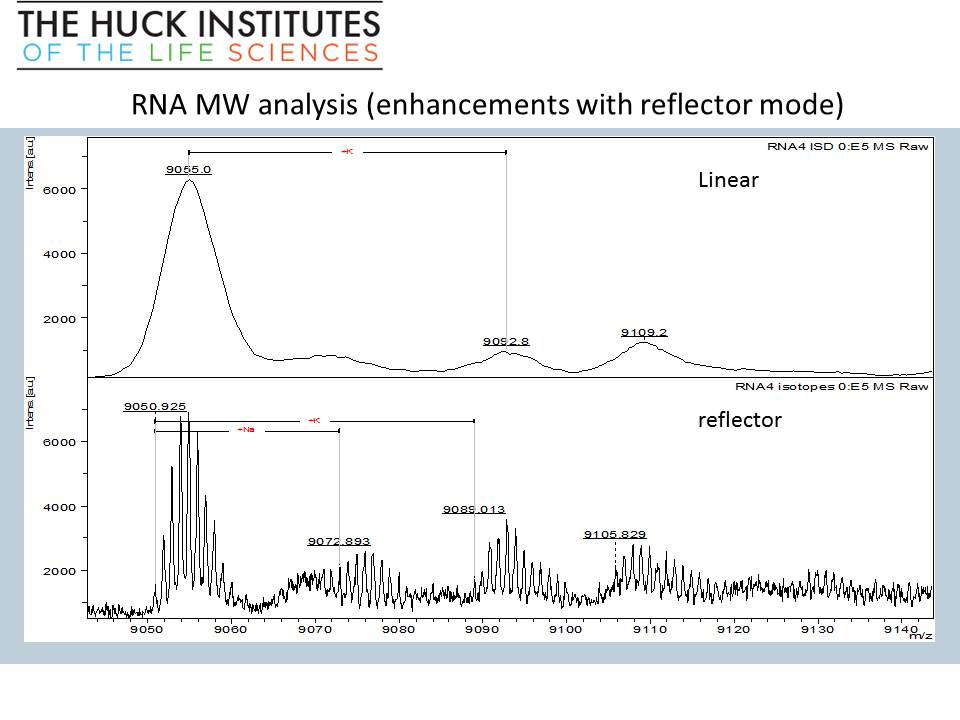
RNA oligomer (28-mer) MW and sequence analysis – on just 1 microliter of sample

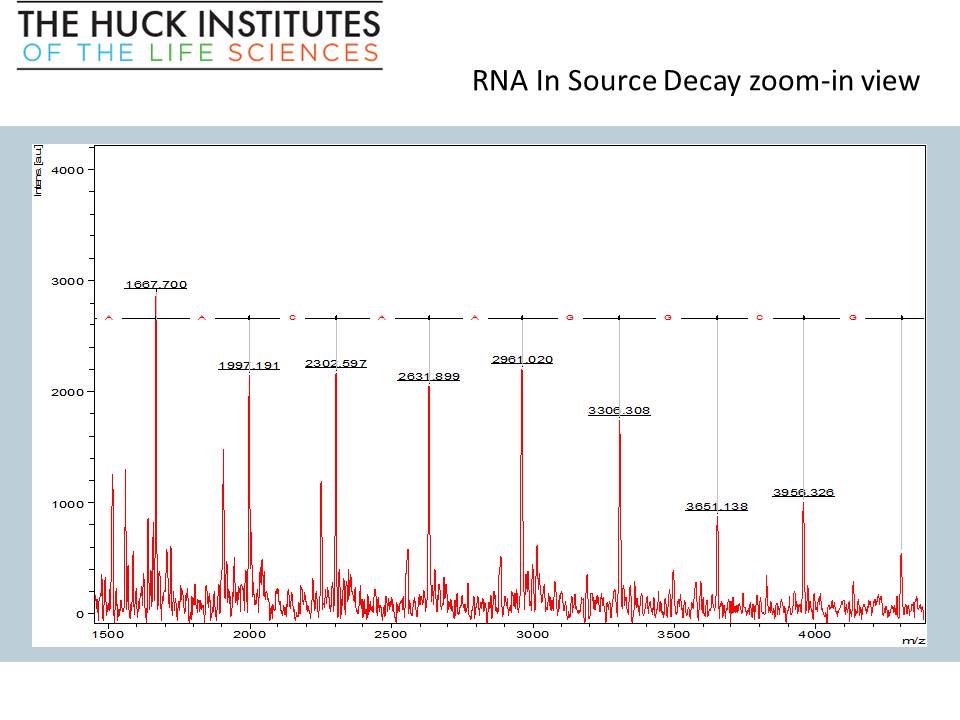

PolyTools software screenshot
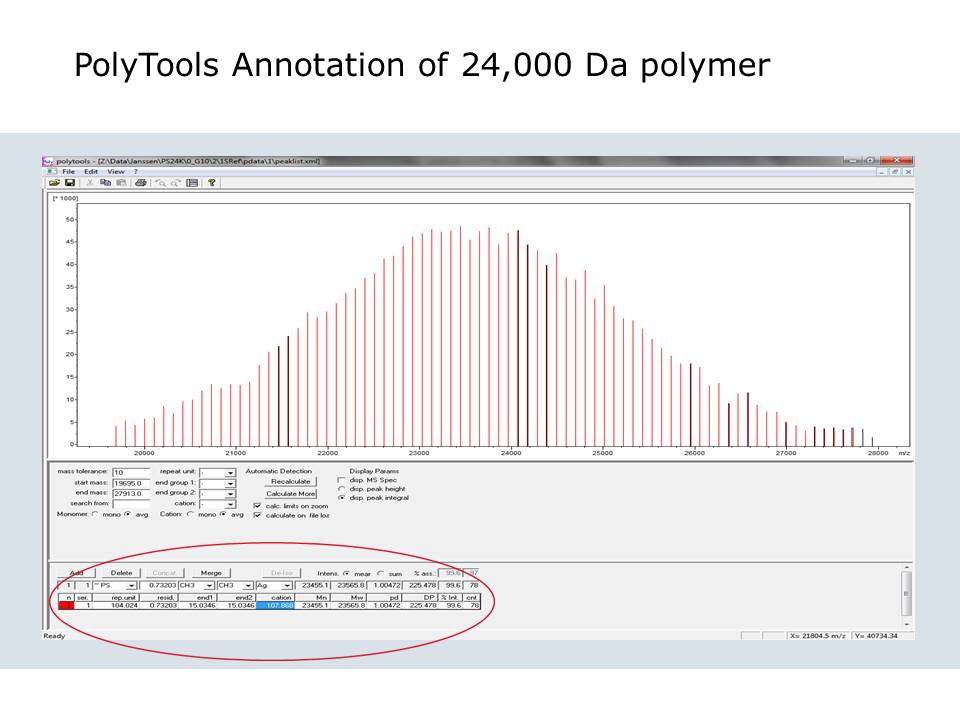
Thank you all for participating and asking questions!
Tutorial: In-gel digestion
If you have never done in-gel protein digestion, this tutorial is for you! The protocol is really simple and does not require any specialized equipment. Even if your gel has been sitting in the fridge for a month or two, it should still work, but no mold, please! First order of business is to choose a band and cut it out. You will need a clean blade, a clean surface (a clean glass plate or a clean transparency sheet), a clean microcentrifuge tube, and a pair of clean gloves. Everything should be clean: the goal is to minimize contamination of interesting proteins by uninteresting keratins.
1. Place the gel on a glass plate, blot excess water with a clean paper tissue. Select a band and cut out only the stained portion of the gel. Avoid unstained area: it will sponge up a protease solution, giving nothing interesting in return.
2. Dice the band into 1-mm (1/16”) sections; this will help the protease reach more protein inside the gel.
3. Place the gel cubes in a clean microcentrifuge tube, cover with water to prevent them from drying out, and label the tube with no more than 5 characters. For example, use your initials and numbers.
4. Include a positive control! There is no charge for analyzing a control! Cut out a known protein band following steps 1, 2, and 3.

The in-gel digestion protocol can be found on Thermo website along with the product numbers for all necessary reagents.
Washing the gel
You will need 50% acetonitrile (ACN) solution containing 8 mg/mL ammonium bicarbonate (wash solution). Add 0.05 – 0.1 mL of this solution per gel band, enough to completely cover the gel. Incubate at 37 C for 15-20 min, discard the solution. Repeat two more times. At this point, all or most of the blue color should be gone. If you are destaining SYPRO, there is no easy way to tell whether it is all gone, of course.
Reducing disulfides and alkylating Cys residues
You will need
- 5 mM TCEP (Tris(2-carboxyethyl)phosphine hydrochloride) solution in the 8 mg/mL ammonium bicarbonate. I make 100 mM (29 mg/mL) stock solution in water and store it in the fridge up to a month. Dilute this stock 20x (e.g. 50 uL 100 mM TCEP + 950 uL ammonium bicarbonate solution).
- Fresh 100 mM IAA (iodoacetamide) solution in the 8 mg/mL ammonium bicarbonate. This is approximately 18 mg/mL IAA solution.
Cover the gel slices with 5 mM TCEP and incubate 10 min at 60 C. Remove the TCEP solution and cover the gel with 100 mM IAA solution. Incubate at 37 C for 15 min with occasional shaking, preferably protected from light. Room temperature will also work, but give it extra 10-15 min. Discard the IAA solution and wash the gel with the wash solution three times to remove IAA and TCEP. (In the Pierce protocol, you might notice that 100 mM IAA requires 9.3 mg in 1 mL, while MW of IAA is 184.9. It is a typo. Either way: 50 mM, 9.3 mg/mL or 100 mM, 18 mg/mL, will work as there will be a large molar excess of IAA compared to Cys. Shrink the gel by covering it with ACN and incubating at r.t. for 15 min or until the gel turns white and brittle. Remove ACN and allow the gel to air dry for 15 min at 37 C (or longer at room temperature).
Digestion
Make 1 mg/mL trypsin stock solution in 50 mM acetic acid or 1 mM hydrochloric acid. This solution can be aliquoted and stored in a freezer for months. Dilute trypsin to 0.01 mg/ml (1:100 dilution) with 8 mg/mL ammonium bicarbonate. Add 50 uL of this solution per gel band and incubate at 37 C 8-24 hours. If you are working with a large piece of gel or several bands combined in one tube, make sure that after the gel re-hydrates in the enzyme solution, it is completely covered. Add more enzyme solution if necessary.
Extracting the peptides
You will need 50% ACN solution containing 0.1% formic acid (FA). If you don’t have formic acid in your lab, let me know – we will share ours with you. Transfer the digest solution to a new tube. Extract each gel band with 50 uL of 50% ACN/0.1% FA (more, if working with a large volume of gel) by incubating at 37 C for 15 min. Transfer this solution to the new tube with the digest solution. Repeat 2 more times. Evaporate the combined extracts in a vacuum concentrator (e.g. Speedvac). Please note, if you are extracting large peptides (>5000 Da), using a sonic bath may be a better option than incubation. Also, in addition to the three 50% ACN extractions, you can use 100% acetonitrile for the final extraction. Submit your dried samples along with a picture of the gel so that I can estimate how much of each sample to use for analysis, and don’t forget to fill out the Protein ID Request form
As always, let me know if you have any questions!