In the current situation (COVID-19 pandemic), a lot has been debated about pooling samples in order to save time, cost, or reagents. It has been suggested that one could pool samples of 32 or 64 samples and test them all at once. If the pooled sample is negative, great — all individual samples are negative. If it’s positive, further re-testing is warranted. Of course, different more or less creative versions of pooling are possible, such as pooling rows and diagonals in order to reduce the total number of tests.
The disadvantage of these approaches is that at least some re-testing is pretty much unavoidable. Thus, ideally, one would pool in a manner that would require no re-testing at all. And here’s where the idea of hashing or chemical bloom filter comes in.
Imagine you split your sample and pool it with other samples. But, you will be smart about how you do this. You will distribute each part of each of your sample on your 96-well plate in a pre-defined manner. That way, based on the combination of positive wells, you could tell exactly which of the original samples were positive. How, you ask? The explanation, code and the whole original idea by Tomáš Janoušek (@Liskni_si) are described here: https://github.com/liskin/covid19-bloom and illustrated below:

When the number of positive samples per 1,000 is low, such as below 1% (less than 10 positive samples per 1,000 tested), the number of false positives is remarkably low. For example, for 8/1000 positive samples, splitting each sample into 5, only single PCR reaction is needed on a 96-well plate to tell you exactly which those 8 positive samples are, and, in this specific scenario, also 1 extra sample that is false positive (although validating all 8+1 samples is not that labor-intensive and could be done).

There won’t be any positive samples you would algorithmically miss (bloom filter doesn’t have false negatives) and also few (if any) false positives.
The caveat is the act of pooling itself; while adding 40 or 50 samples to a single tube is definitely easy for a robot, it could be error-prone when done manually. While a human could pool rows or columns relatively easily, asking someone to pipette 1,000 samples, and moreover each of those into 3 wells, more so in a pre-defined manner, might simply be too much of an ask. Reducing the number of tested samples to 500 and capping the number of samples in a tube will generate the following plot:
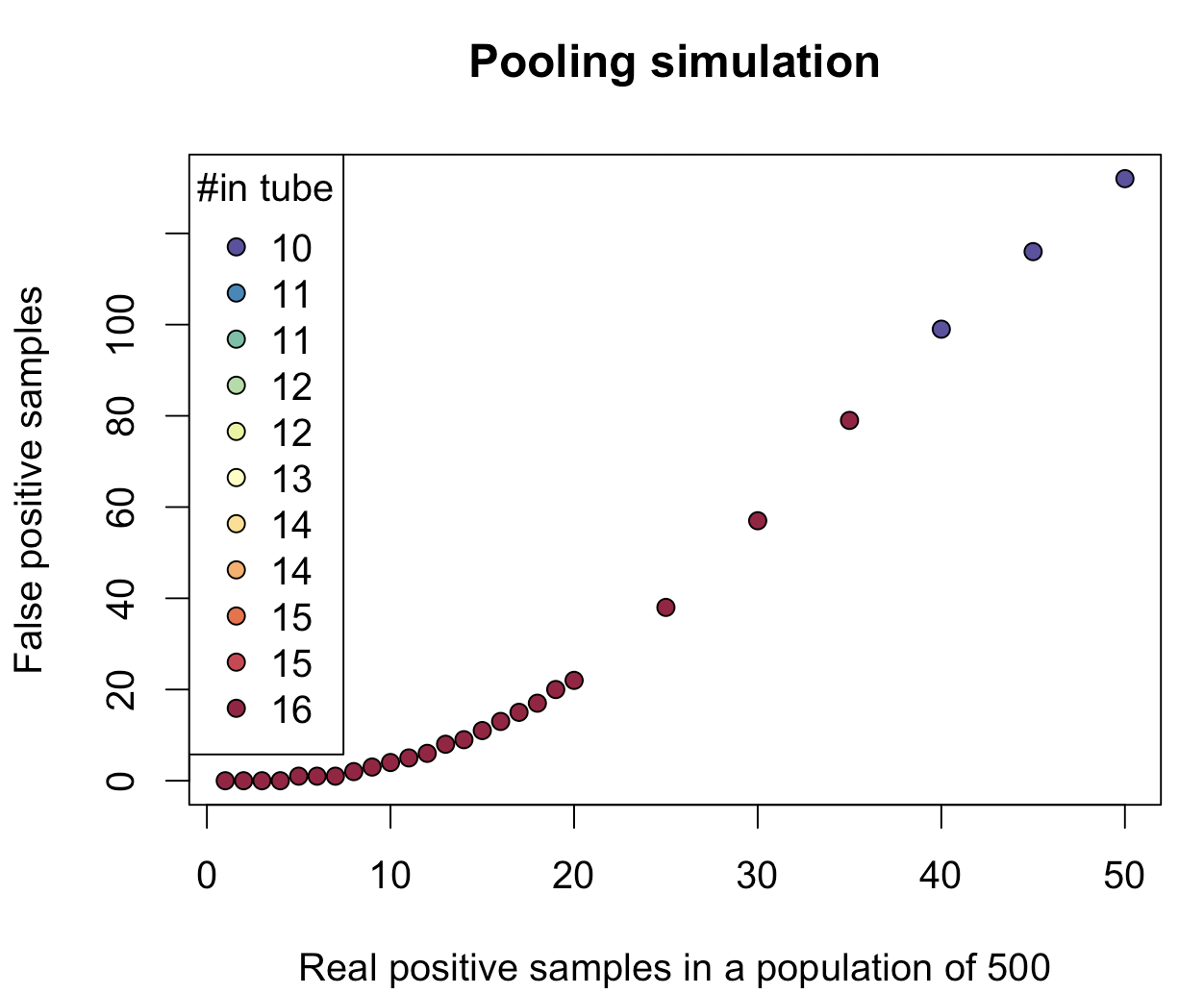
Here, all 500 samples are separated into three pieces, and the number of real positives versus false positives is plotted, while keeping the total number of pooled samples in a tube low.
This all brings us to the discussion of the next bottleneck: RNA extraction. In all this pooling, what do we actually pool? Extracted RNA? If that’s the case, we still need to do 1,000 extractions for 1,000 samples. And then split those 1,000 extractions into 2, 3, 4 or 5 parts (a parameter that can be chosen). And we know that with all this testing, the extraction kits are running low.
We also know that with the progress of the disease a viral load in a patient changes — someone who might have tested negative yesterday could be positive today. So when a swab is taken, a viral load might be low to begin with, without any further amplification step. However, we do amplify, over many cycles. So perhaps it doesn’t matter if we start with 1/3rd or 1/4th of the original load. This means that if a swab could be mechanically split, with the sufficient viral load still there, one could pool these parts of the original swab.
Thus, for 1,000 samples, only 96 RNA extractions are needed. And we can still confidently say which individual samples are positive.
In conclusion, for a population of samples with a small percentage of positives, the chemical bloom filters could represent a fast and efficient way to reduce the number of performed tests and save reagents (1,000 samples can be tested using 96 pooled tubes).
The data for the two plots above can be found below:
nRealPositive nAll kHashes samplesInTube avgFalsePositives 1 1000 4 41.67 0.00 2 1000 4 41.67 0.01 3 1000 5 52.08 0.03 4 1000 5 52.08 0.07 5 1000 5 52.08 0.19 6 1000 5 52.08 0.40 7 1000 5 52.08 0.78 8 1000 5 52.08 1.29 9 1000 5 52.08 2.12 10 1000 5 52.08 3.23 11 1000 5 52.08 4.97 12 1000 5 52.08 7.25 13 1000 5 52.08 10.00 14 1000 5 52.08 13.27 15 1000 5 52.08 17.13 16 1000 4 41.67 21.30 17 1000 4 41.67 25.41 18 1000 4 41.67 29.96 19 1000 4 41.67 35.00 20 1000 4 41.67 40.67 25 1000 3 31.25 75.94 30 1000 3 31.25 114.93 35 1000 3 31.25 160.06 40 1000 2 20.83 208.18 45 1000 2 20.83 244.67 50 1000 2 20.83 279.05
nRealPositive nAll kHashes samplesInTube avgFalsePositives 1 500 3 15.62 0.00 2 500 3 15.62 0.04 3 500 3 15.62 0.17 4 500 3 15.62 0.36 5 500 3 15.62 0.56 6 500 3 15.62 0.93 7 500 3 15.62 1.44 8 500 3 15.62 2.14 9 500 3 15.62 2.96 10 500 3 15.62 3.95 11 500 3 15.62 5.07 12 500 3 15.62 6.32 13 500 3 15.62 7.64 14 500 3 15.62 9.18 15 500 3 15.62 10.99 16 500 3 15.62 12.96 17 500 3 15.62 15.08 18 500 3 15.62 17.34 19 500 3 15.62 19.80 20 500 3 15.62 22.37 25 500 3 15.62 37.95 30 500 3 15.62 57.12 35 500 3 15.62 79.27 40 500 2 10.42 98.58 45 500 2 10.42 115.78 50 500 2 10.42 131.52
Disclaimer: We are not epidemiologists and we are publishing this with an intent to induce further discussions and improvements. Our conclusions are not intended as public-health advice and shouldn’t be regarded as such.





{kind=link}