
DNA damage is a major determinant of cell fate and thus poses a significant threat to organism health. On one hand, incorrect repair or replication of damaged DNA can result in accretion of mutations, referred to as genomic instability, which is considered an enabling characteristic of cancer. On the other hand, cancer therapy such as radiation and genotoxic chemotherapy, works by inducing lethal DNA damage in cancer cells. At the molecular level, DNA damaging agents cause DNA lesions or adducts that affect on or both strands of the DNA molecule. In proliferating cells, unrepaired DNA damage poses a major threat to genome stability as encountering DNA lesions arrests the progression of the high-fidelity replicative DNA polymerases. Prolonged stalling of the replication fork at these sites can lead to fork collapse and formation of double stranded DNA breaks (DSBs) which can cause cell death or initiate chromosomal translocations. To avoid this, fork stalling initiates a set of complex molecular events aimed at bypassing the lesion to quickly restore normal replication, without technically repairing the damage which is left behind to be fixed at a later time. At least three strategies have been described for restarting stalled forks (Fig. 1). Translesion synthesis (TLS) involves a switch from the replicative polymerase to one of the many low-fidelity TLS polymerases which can insert nucleotides across DNA lesions. This process is frequently mutagenic, as the original base on the template strand is unreadable by the TLS polymerases. To keep this mutagenic process under control, TLS is regulated so that it only occurs upon spatially and temporary restricted post-translational modification of the replication fork protein PCNA, which is a co-factor for replicative polymerases in its unmodified form, and for TLS polymerases in its mono-ubiquitinated form. Alternatively, in an error-free process, stalled forks can be restarted upon template switching (TS) which involves their regression by translocases such as ZRNAB3, annealing the two nascent strands, and allowing replication restart using the nascent strand of the unaffected sister chromatid as template. TS requires poly-ubiquitination of PCNA with K63-linked ubiquitin chains, which provide a docking platform for ZRANB3. Finally, a PCNA-independent process is also available, through homologous recombination (HR)-mediated engagement of the sister chromatid by the BRCA-RAD51 recombinase complex. The decision making process that regulates which one of this mutually-exclusive pathways is employed is not understood.
Our vision is to provide novel drug targets for cancer prevention and personalized cancer treatment. To achieve this long-term goal, our laboratory employs a combination of cellular, molecular, genetic, genome editing, and functional genomics methods. Our research program encompasses three strategic themes:
- To understand the molecular machineries involved in maintaining genomic integrity during DNA replication.
- To uncover novel mechanisms of repair of DNA damage induced by radiation, chemotherapy, and environmental agents.
- To infer how the impact of individual tumor genetic make-up on the response to cancer therapy

Current and past research projects
1. PCNA and its post-translational modifications
PCNA is a homotrimeric ring-shaped protein that encircles DNA, conferring processivity to DNA polymerases during DNA replication. Post-translational modification of PCNA by ubiquitin and SUMO regulates fork restart and lesion bypass through mutagenic and non-mutagenic processes.



- Figure 2. PCNA and its modifications. A. Model of DNA replication showing PCNA at the replication fork (from Burges, 2009.) B. Front and side view of PCNA. Arrows indicate Lys 164. C. PCNA modifications: ubiquitination signals lesion bypass by translesion synthesis or template switching; SUMOylation inhibits HR by recruiting PARI (in mammals) or Srs2 (in yeast) (adapted from Bergink and Jentsch, 2009).
We recently identified an unexpected role of PCNA ubiquitination in maintaining replication fork protection (Thakar et al, Nature Communications 2020). We found that, under unperturbed growth conditions, PCNA ubiquitination is required for efficient lagging strand synthesis by mediating gap-filling behind progressing replication forks. In the absence of PCNA ubiquitination, persistent gaps are formed between the lesion and the previous Okazaki fragment. These gaps interfere with Okazaki fragment maturation and subsequent PCNA unloading by ATAD5. By sequestering the CAF-1 chromatin assembly complex, PCNA retention on the lagging strand alters the efficiency of chromatin establishment which results in replication forks encountering a sparse chromatin organization. Upon fork arrest and reversal, the abnormal structure generated is a substrate for uncontrolled resection by DNA2.

- Figure 3. Schematic representation of the proposed role of the newly-identified UbiPCNA–LIG1–ATAD5–CAF-1 pathway in replication fork protection (from Thakar et al, Nature Communications 2020).
2. Functional genomics studies using CRISPR genome-wide libraries to uncover genetic vulnerabilities for cancer treatment.
Personalized cancer treatment involves identification of genetic vulnerabilities in individual patients’ tumors, which confer an improved response to therapy. Using whole-genome CRISPR libraries, we can suppress or activate the expression of each of the approx. 20,000 human genes, and investigate how that genetic modification impacts the cell’s response of the cell to a drug. For example, we characterized the genetic determinants that confer sensitivity or resistance to the PARP inhibitor drug olaparib, used in the clinical treatment of BRCA-mutated ovarian tumors (Clements et al, Nature Communications 2020). We also explored the genetic landscape controlling the resistance to ATR inhibition (Schleicher et al, PLoS Genetics 2020) and PARP14-synthetic lethality (Dhoonmoon et al, Nucleic Acids Research 2020).

- Figure 4. Schematic representation of the CRISPR knockout screen for olaparib resistance in HeLa BRCA2-knockout cells. Cells were infected with the Brunello CRISPR knockout library. Infected cells were divided into PARP inhibitor (olaparib)-treated or control (DMSO) arms. Genomic DNA was extracted from cells surviving the drug treatment and single-guide RNAs (sgRNAs) were identified using Illumina sequencing (from Clements et al, Nature Communications 2020).
3. Novel mechanisms of fork protection and chemoresistance in BRCA-mutant cells.
Suppression of nascent DNA degradation has emerged as an essential role of the BRCA pathway in genome protection. In BRCA-deficient cells, the MRE11 nuclease is responsible for both resection of reversed replication forks, and accumulation of single stranded DNA gaps behind forks. We recently showed that the mono-ADP-ribosyltransferase PARP14 is a critical co-factor of MRE11 (Dhoonmoon et al, Nature Communications 2022). PARP14 is recruited to nascent DNA upon replication stress in BRCA-deficient cells, and through its catalytic activity, mediates the engagement of MRE11. Loss or inhibition of PARP14 suppresses MRE11-mediated fork degradation and gap accumulation, and promotes genome stability and chemoresistance of BRCA-deficient cells. Moreover, we showed that the KU complex binds reversed forks and protects them against EXO1-catalyzed degradation. KU recruits the PARP14-MRE11 complex, which initiates partial resection to release KU and allow long-range resection by EXO1. Our work identified a multistep process of nascent DNA processing at stalled replication forks in BRCA-deficient cells.

- Figure 5. KU binds the exposed DSB end of symmetrical reversed forks, protecting it against EXO1. At the same time, KU bound on the reversed fork recruits the PARP14-MRE11 complex, and through its endonuclease activity MRE11 creates a nick, which is then process by its 3’ to 5’ exonuclease activity towards the DSB end. This results in release of KU from the DSB end. In BRCA-proficient cells, loading of RAD51 stabilizes the ssDNA overhang against further nucleolytic processing. In the absence of KU, EXO1 engages the DSB end with its 5’ to 3’ exonuclease activity for, but loading of RAD51 by the BRCA pathway on the partially resected DSB end stabilizes it against further degradation. In BRCA-deficient cells, the partially resected structure is susceptible to continuous (long-range) degradation by EXO1 on the 5’ end strand, and MRE11 (and potentially other 3’ to 5’ exonucleases) on the 3’ end strand (model from Dhoonmoon et al, Nature Communications 2022).
Another hallmark of BRCA-mutant cells recently emerged is the inability to suppress replication-associated single-stranded DNA (ssDNA) gaps. We recently reported that lagging strand ssDNA gaps interfere with the ASF1-CAF-1 nucleosome assembly pathway, and drive fork degradation in BRCA-deficient cells (Thakar et al, Nature Communications 2022). We showed that CAF-1 function at replication forks is lost in BRCA-deficient cells, due to defects in its recycling during replication stress. This CAF-1 recycling defect is caused by lagging strand gaps which preclude PCNA unloading, causing sequestration of PCNA-CAF-1 complexes on chromatin. Importantly, correcting PCNA unloading defects in BRCA-deficient cells restores CAF-1-dependent fork stability. We further showed that the activation of a HIRA-dependent compensatory histone deposition pathway restores fork stability to BRCA-deficient cells. We thus defined lagging strand gap suppression and nucleosome assembly as critical enablers of BRCA-mediated fork stability.

- Figure 6. The failure of BRCA-deficient cells to restrain fork progression during replication stress causes gap accumulation due to repriming by PRIMPOL (on the leading strand) and Polα (on the lagging strand). Polα-mediated repriming results in formation of lagging strand gaps which preclude PCNA unloading. The persistence of PCNA behind replication forks sequesters CAF-1 away from active replication factories, thus interfering with nucleosome assembly at forks and priming them for degradation upon stalling (model from Thakar et al, Nature Communications 2022).
4. Processing of single stranded DNA gaps at stressed replication forks.
Accumulation of single stranded DNA (ssDNA) gaps in the nascent strand during DNA replication has been associated with cytotoxicity and hypersensitivity to genotoxic stress, particularly upon inactivation of the BRCA tumor suppressor pathway. However, how ssDNA gaps contribute to genotoxicity is not well understood. We recently described a multi-step nucleolytic processing of replication stress-induced ssDNA gaps which converts them into cytotoxic double stranded DNA breaks (DSBs) (Hale et al, Nature Communications 2023). We showed that ssDNA gaps are extended bidirectionally by MRE11 in the 3’-5’ direction and by EXO1 in the 5’-3’ direction, in a process which is suppressed by the BRCA pathway. Subsequently, the parental strand at the ssDNA gap is cleaved by the MRE11 endonuclease generating a double strand break. We also show that exposure to bisphenol A (BPA) and diethylhexyl phthalate (DEHP), which are widespread environmental contaminants due to their use in plastics manufacturing, causes nascent strand ssDNA gaps during replication. These gaps are processed through the same mechanism described above to generate DSBs. Our work sheds light on both the relevance of ssDNA gaps as major determinants of genomic instability, as well as the mechanism through which they are processed to generate genomic instability and cytotoxicity.

- Figure 7. The sequential activities of EXO1 and MRE11 exonuclease activities and or MRE11 endonuclease activity in ssDNA gap formation. The MRE11 3’-5’ exonuclease activity and the EXO1 5’-3’ exonuclease activity bidirectionally expand a short ssDNA gap. Subsequent to exonucleolytic gap expansion, the MRE11 endonuclease cleaves the template strand to cause a double stranded DNA break (model from Hale et al, Nature Communications 2023).
5. The role of mono-ADP-ribosylation in DNA repair.
Posttranslational modifications are widely used to regulate DNA repair processes. Poly-ADP-ribose polymerization is a type of post-translational modification catalyzed by PARP enzymes, with important roles in repair of single and double strand breaks. While the functions of PARP1 are well characterized, there are over 17 PARPs in human cells and most of them have not been characterized. Only recently it was shown that a subset of PARPs have mutations in the active site that abolishes poly-ADP-ribosyltranferase activity; instead, these enzymes can only catalyze the attachment of a single ADP-ribose molecule, a process termed mono-ADP-ribosylation. The role of this modification was not known, and in particular, it had not been associated with DNA repair previously. Our laboratory is the first to show that mono-ADP-ribosylation plays an essential role in DNA repair. We showed that the mono-ADP-ribosyltransferase PARP10 interacts with the PCNA, the master regulator of DNA replication, to promote replication of damaged DNA and DNA damage tolerance (Nicolae et al, J. Biol. Chem. 2014). We also found that the related ADP-ribosyltransferase PARP14 promotes double strand break repair by Homologous Recombination, through mono-ADP-ribosylation of RAD51 (Nicolae et al, Nucleic Acids Research 2015). Moreover, we recently showed that PARP10 mutation causes neurodegeneration in humans (Shahrour et al, Neurogenetics 2016). Finally, we found that PARP10 plays an essential role during carcinogenesis (Schlacher et al Nucleic Acids Research 2018). Our work described for the first time a previously unanticipated role for mono-ADP-ribosylation in DNA repair, and uncovered novel biological functions for the poorly characterized enzymes PARP10 and PARP14.

- Figure 8. PARP10 and PARP14 regulate PCNA-dependent fork restart. DNA damaging carcinogens (such as benzo[a]pyrene from tobacco smoke) form DNA lesions that block the progression of replicative DNA polymerases. Outcomes of fork arrest include both point mutations and chromosomal translocations, depending how fork restart mechanisms are engaged. Bypass of DNA adducts can occur through Translesion Synthesis (TLS), which employs specialized polymerases able to replicate through DNA lesions. Due to their intrinsically low fidelity, incorrect nucleotides can be inserted across the lesion. Upon arrest of the replication machinery at a DNA adduct, PCNA becomes ubiquitinated to specifically recruit TLS polymerases containing ubiquitin-recognition motifs. PARP10 regulates PCNA-dependent TLS. If the adduct is not bypassed, fork collapse can occur, resulting in DNA strand breaks. The Homologous Recombination (HR) machinery can use the sister chromatid to re-establish the replication fork. HR is generally error-free, and it is an essential mechanism protecting against carcinogenesis. Binding of the protein RAD51 to broken DNA is an essential step in the HR reaction, and we showed that PARP14 regulates this step.
6. Unraveling the mechanism of DNA damage-induced differentiation of myeloid leukemia cells.
DNA damage exposure is a major modifier of cell fate in both normal and cancer tissues. In response to DNA damage, myeloid leukemia cells activate a poorly understood terminal differentiation process. We found that the NFkB pathway directly activates expression of the proliferation inhibitor p21 in response to DNA damage in myeloid leukemia cells (Nicolae et al, Oncogene 2018). In order to understand the role of this unexpected regulatory event, we ablated the NFkB binding site we identified in the p21 promoter, using CRISPR/Cas9-mediated genome editing. We found that NFkB -mediated p21 activation controls DNA damage-induced myeloid differentiation. Moreover, we showed that PARI, a replisome protein involved in regulating DNA repair and replication stress, is overexpressed in myeloid leukemia cells, and its knockdown reduces leukemia cell proliferation in vitro and in vivoin xenograft mouse models (Nicolae et al, Oncogene 2019). PARI depletion enhances replication stress and DNA damage accumulation, coupled with increased myeloid differentiation. Mechanistically, we showed that PARI inhibits activation of the NFkB pathway. Finally, we showed that PARI expression negatively correlates with expression of differentiation markers in clinical myeloid leukemia samples, suggesting that targeting PARI may restore differentiation ability of leukemia cells and antagonize their proliferation. Our results uncover a p53-independent pathway for p21 activation involved in controlling hematopoietic cell fate.
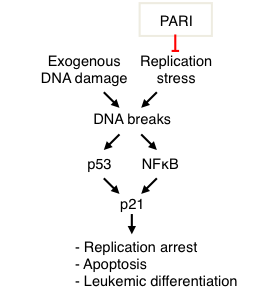
- Figure 9. PARI suppresses replication stress in myeloid leukemia cells, which would otherwise result in DNA damage accumulation. DNA damage can activate the NFkB pathway to promote p53-independent expression of p21, resulting in cell cycle arrest and myeloid differentiation (from Nicolae et al, Oncogene 2018).
7. Novel mechanisms of PARP inhibitor resistance.
BRCA proteins are essential for Homologous Recombination DNA repair, and their germline or somatic inactivation is frequently observed in human tumors. Understanding the molecular mechanisms underlying the response of BRCA-deficient tumors to chemotherapy is paramount for developing improved personalized cancer therapies. While PARP inhibitors have been recently approved for treatment of BRCA-mutant breast and ovarian cancers, not all patients respond to this therapy, and resistance to these novel drugs remains a major clinical problem. Several mechanisms of chemoresistance in BRCA2-deficient cells have been identified. Rather than restoring normal recombination, these mechanisms result in stabilization of stalled replication forks, which can be subjected to degradation in BRCA2-mutated cells. We recently identified the first mechanism of resistance to PARP inhibitors which restores recombination in BRCA2-deficient cells (Clements et al, Nucleic Acids Research 2018). We showed that the transcriptional repressor E2F7 modulates the chemosensitivity of BRCA2-deficient cells. We found that BRCA2-deficient cells are less sensitive to PARP inhibitor and cisplatin treatment after E2F7 depletion. Moreover, we showed that the mechanism underlying this activity involves increased expression of RAD51, a target for E2F7-mediated transcriptional repression, which enhances both Homologous Recombination DNA repair, and replication fork stability in BRCA2-deficient cells. Our work describes a new mechanism of therapy resistance in BRCA2-deficient cells, and identifies E2F7 as a putative biomarker for tumor response to PARP inhibitor therapy.
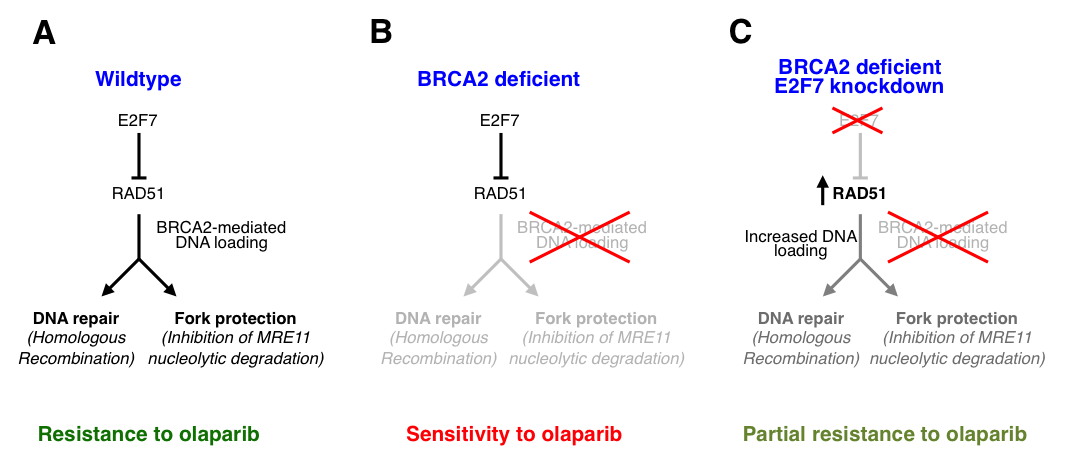
- Figure 10. Model showing the impact of E2F7 on olaparib sensitivity of BRCA2-deficient cells. In wildtype cells, BRCA2-dependent loading of RAD51 to DNA breaks and stalled replication forks ensures correct HR and fork protection, which promote resistance to olaparib (A). In BRCA2-deficient cells, RAD51 loading is reduced and both HR and fork protection are impaired, which results in olaparib sensitivity (B). E2F7 represses expression of RAD51. Loss of E2F7 results in increased RAD51 levels, which allows more binding of RAD51 to damaged DNA. This promotes both HR and fork protection, restoring resistance to olaparib (C) (from Clements et al, Nucleic Acids Research 2018).
8. Impact of germline mutations in genome stability factors on human health.
While inherited deficiencies in DNA repair genes have been indisputably linked to cancer predisposition, the impact of DNA and chromatin stability factors on organism development in humans is less clear. In collaboration with Dr. Orly Elpeleg (Hadassah Medical Center, Israel) we showed that germline mutations in such factors, including PARP10 (Shahrour et al, Neurogenetics 2016), UBTF (Edvardson et al, American Journal of Human Genetics 2018), and RNF13 (Edvardson et al, American Journal of Human Genetics 2019) in human individuals are associated with severe neurodevelopmental disorders. These findings underscore the essential role of genome and chromatin stability during development.
9. Novel genomic methods to investigate structural genomic instability
While deep next generation sequencing is ideal for identifying point mutations, structural, translocations and other structural variations are difficult to detect using this technology since the reduced size of the reads (50-200bp) makes it statistically unlikely to chance upon translocation breakpoints. In contrasty, optical genome mapping using the Bionano Saphyr platform allows mapping of megabase-sized DNA, based on imaging of DNA fibers labeled with sequence-specific fluorescent tags. We recently employed this technology to investigate structural genomic variation in myeloid leukemia patients. (Xu et al, BioRvix 2019). We are currently using this approach to investigate mechanisms of genomic instability induced by DNA damage exposure.
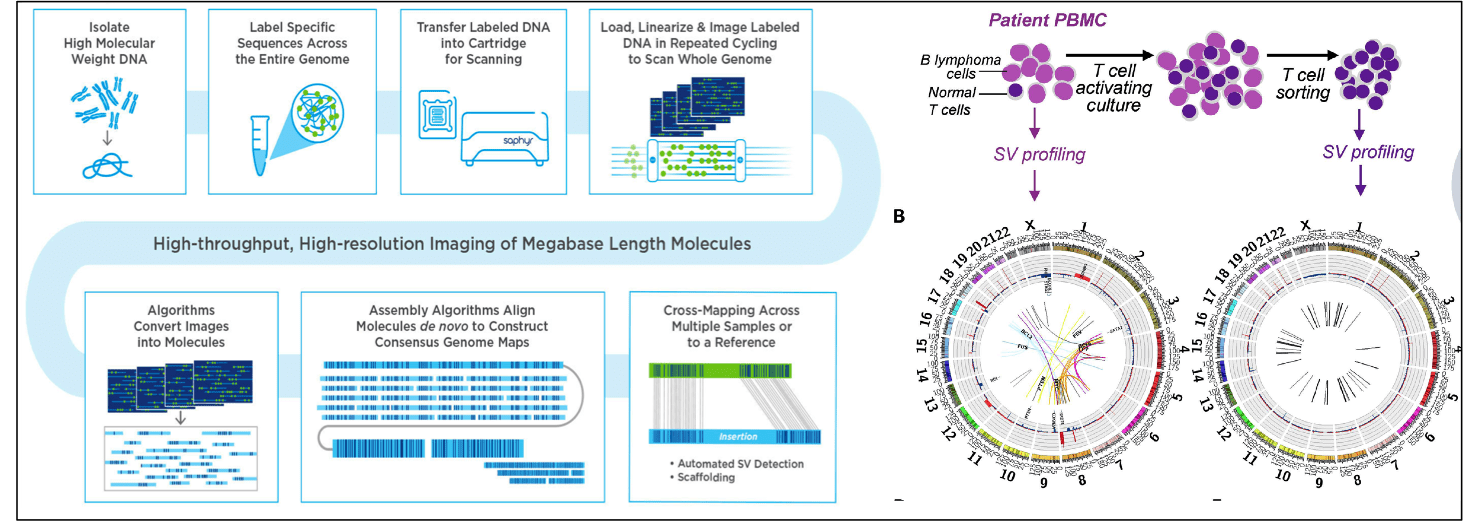
- Figure 11. Workflow for SV detection by optical mapping using the BionanoSaphyrplatform. Samples from a myeloid leukemia patient show previously undetected structural variations, which are missing in matched normal cells (from Xu et al, BioRvix 2019).
9. HUWE1 and replication stress.
We recently found that the HUWE1 ubiquitin ligase is recruited to stalled replication forks by the replication factor PCNA. We found that HUWE1 contains a PCNA interacting domain (PIP-box) required for PCNA interaction. HUWE1-knockout cells show increased replication stress, which can be corrected by re-
expression of wild-type, but not PCNA interaction-deficient HUWE1 mutant. Importantly, we found that HUWE1 mono-ubiquitinates H2AX to promote gH2AX signaling, recruitment of repair proteins, and restart of stalled replication forks. Our work unexpectedly uncovered HUWE1 as a novel E3 ubiquitin ligase for H2AX, responsible for the efficient response to replication stress (Choe et al, EMBO Reports 2016).
- Figure 12. HUWE1 is recruited by PCNA to stalled replication forks to ubiquitinate H2AX (adapted from Coleman and Huang, News and Views, EMBO Reports 2016.)
Research Grants Support:
2016-2026 NIH – NIEHS 2R01ES026184 (PI: Moldovan)
2023-2026 Four Diamonds Transformative Patient-Oriented Cancer Research Project Award (MPI: Moldovan, Nicolae))
2019-2023 NIH –NIGMS 1R01GM134681 (MPI: Bielinsky, Moldovan)
2022-2023 Forma Therapeutics, industry contract
2019-2020 Pennsylvania Dept. of Health TSF CURE award
2015-2020 St. Baldrick Scholar Award from the St. Baldrick Foundation for Cancer Research
2018-2019 4Diamonds Pediatric Cancer Collaborative Research Award
2015-2018 Department of Defense (DoD) Peer Reviewed Cancer Research Program (PRCRP) Career Development Award
2014-2016 Conquer Cancer Award from the Concern Foundation for Cancer Research
2013-2015 V Scholar Award from the Jimmy V Foundation for Cancer Research
2014-2015 Jake Gittlen Foundation Collaborative Cancer Research Award
Collaborating labs:
Anja Bielinsky lab at UVA: R01GM134681 (2019-2023) The Role of DNA damage tolerance pathways in human cells.
Claudia Nicolae lab at Penn State: R01CA244417 (2021-2026) The role of PARP10 in alleviating replication stress and promoting cellular proliferation and tumorigenesis.
Tom Spratt lab at Penn State: R01ES021762 (2018-2023) Genotoxicity and Repair of Tobacco-Specific Nitrosamine DNA Adducts.
Matt Swulius lab at Penn State: R01NS126448 (2023-2028) Using cryo-electron tomography and live-cell fluorescent imaging to study the role of cofilin in regulating neuronal filopodial structure and dynamics.
Greg Yochum lab at Penn State: R03CA279861 (2023-2025) Wnt/beta-catenin signaling in early-onset colorectal cancer.