Recently I wanted to present a table with information from various sources – some blast results, some repeat masker results, some custom-made scripts’ outputs. Cool. Let’s just put it all in Excel, do some conditional formatting and make it pretty. All I need to do is to combine multiple tables into one big nice table, so CTRL+C and CTRL+V will just do the job. An hour later, I felt like this paperclip:

from @tim_yates https://mobile.twitter.com/tim_yates/status/367297797709504513?p=v
Issue is, some tables miss some of the values. Some tables are sorted in one fashion and others in the other, based on how naturally or numerically whoever sorted them felt. So, let’s forget Excel and just join them with proper tools. For the beginning, let’s join two tables, each with 2 columns separated by tab.
join -t $'\t' -a 1 table1 table2
This simple command joins two sorted (e.g. with sort -b) tables based on common column (usually first column which is default behavior). It uses tabulator as separator. The command above is INNER JOIN, only those rows will be reported for which there is entry in both files.
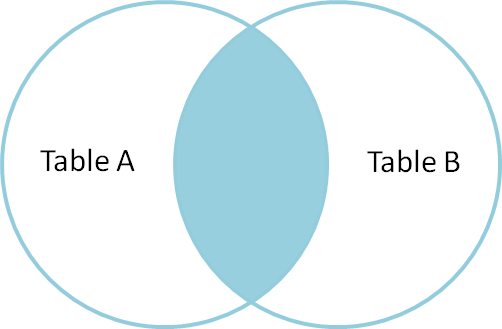
from: http://blog.codinghorror.com/a-visual-explanation-of-sql-joins/
In my case, table2 has less rows than table1, but I want to produce all rows from table1. What I need is LEFT OUTER JOIN. Whenever there is no corresponding value available in table2, empty string will be produced.
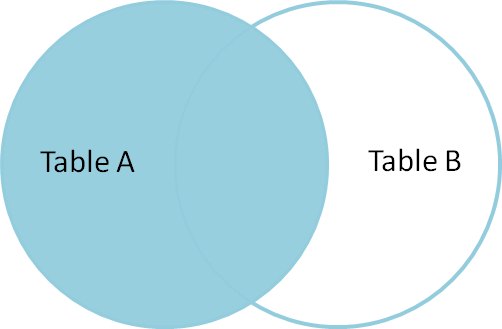
from: http://blog.codinghorror.com/a-visual-explanation-of-sql-joins/
This can be achieved by parameter -a, in our case -a 1 (first file).
-a file_number In addition to the default output, produce a line for each
unpairable line in file file_number [man join]
Empty strings are those of less favorite strings for most people, therefore let’s now use NAN instead of empty strings for all missing values.
join -t $'\t' -a 1 -e "NAN" table1 table2
Nothing happened. Still, only empty strings are reported instead of desired “NAN”. It turns out you need to be specific about which output you want:
join -t $'\t' -a 1 -e "NAN" -o 0,1.2,2.2 table1 table2
In this simple scenario, 0 will output common column (the one we are using for joining) and 1.2 and 2.2 will output second column of table 1 and table 2, respectively.
vigilance needed: 1/10
Happy joining.
Resources:
A Visual Explanation of SQL Joins; http://blog.codinghorror.com/a-visual-explanation-of-sql-joins/
join – relational database operator; http://pubs.opengroup.org/onlinepubs/007904975/utilities/join.html