
Introduction
Hydrogen atoms on Cu(111) can bind in two distinct threefold sites, HCP and FCC. This post is aimed at showing energy difference between these two sites without and with zero point energy correction by using DFT calculation.
For harmonic oscillator, in classical mechanics, the energy for equilibrium state is zero kinetic energy plus potential energy E0; But in quantum mechanics, due to Heisenburg uncertainty principle, the energy changes to: \begin{equation} E = E_0 + \Sigma_i \frac{h \nu_i}{2} \end{equation}
E0 is still the potential energy and ν is the vibrational frequency of the oscillator. Zero point energy is defined as: \begin{equation} E_{ZPE} = \Sigma_i \frac{ h \nu_i}{2} \end{equation}
Normally, zero point energy is not trivial for light atoms, like H.
In order to get zero point energy, vibration frequencies of H on Cu surface are needed and these frequencies are calculated from mass-weighed Hessian Matrix A, which has a relation with Hessian matrix H:
\begin{equation} A_{ij} = H_{ij} / m_i \end{equation}
where mi is mass of the atom associated with ith coordinate. In this post m =1.00794*1.66054*10^-27 kg. The eigenvalues of matrix A λ could give vibration frequencies by: \begin{equation} \nu_i = \sqrt{\lambda}/2\pi \end{equation}
Constructing Hessian Matrix requires us to do second-order differentiation for energy to coordinate: \begin{equation} H_{ij} = (\frac{\partial E}{\partial x_i \partial x_j})_{x=0} \end{equation}
which could be obtained from finite difference approximation. The method used in this post is the simple one which involving three points in one differentiation:
\begin{equation} H_{ij} \simeq \frac{E(\delta x_i, \delta x_j) – 2 E_0 + E(- \delta x_i, – \delta x_j)}{\delta x_i \delta x_j} \end{equation}
The model built in this post is H on the Cu(111) surface with three layers, vacuum above the surface is 10 Å. 2*2 supercell is adopted in case the hydrogen atoms in periodic structure are too close.
Before individual energy calculations, geometry optimization is conducted first. The hydrogen atom is put near the HCP site (or FCC site). Bottom two layers are fixed and the first layer and hydrogen are allowed to relax. After geometry optimization is finished, the distance between H atom and nearest three Cu atom could be checked. Fig. 1, 2 and 3 show the diagrams of the model. Fig. 1 shows that after geometry optimization, the distances between H atom and nearest three Cu atoms are close. So does Fig. 2. Fig.3 shows the side-view of the model.
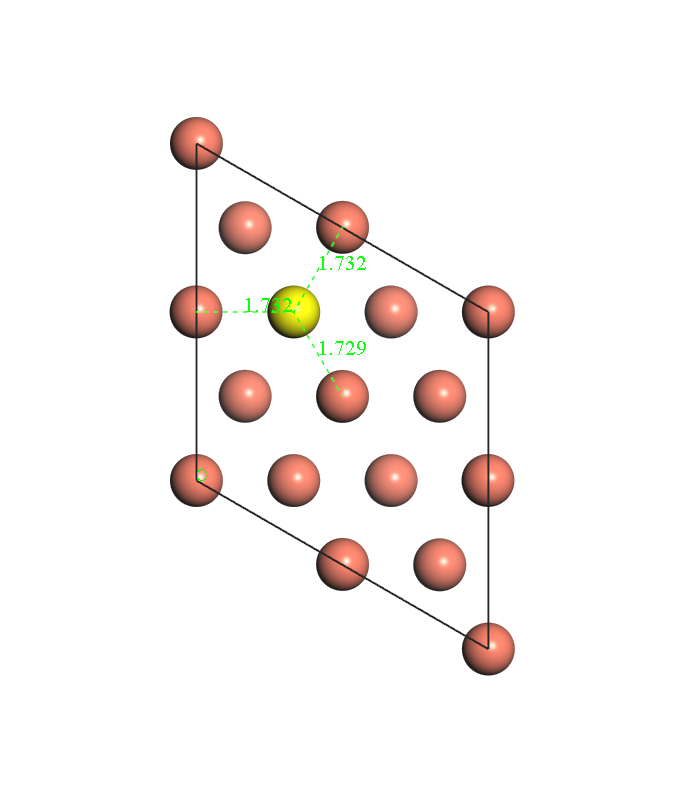
Fig. 1 diagram for HCP site after geometry optimization
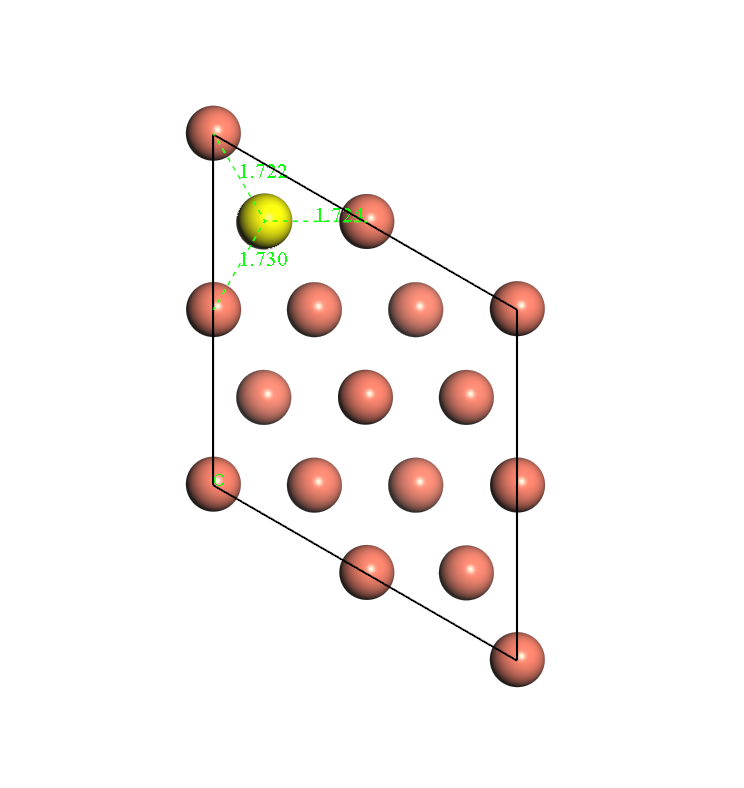
Fig. 2 diagram for FCC site after geometry optimization

Fig. 3 side-view of ‘H on Cu(111)’ model
Based on the geometry optimized, H atom is moved to three directions (x, y, z) according to elements in Hessian Matrix and energy calculation is conducted after each movement. The move step is 0.04Å.
Functional: GGA PBE[1]; k points: 5*5*1 ; cutoff energy: 408.2 eV; SCF tolerence: 2.0*10^-6 eV/atom; Pseudopotentials: OTFG ultrasoft, for Cu: inner shell is 1s2 2s2 2p6 3s2 3p6, outer shell is 3d10 4s1, for H: outer shell is 1s1; dipole correction is included since the model is not symmetric.
(‘CASTEP’ calculation module[2] is adopted for all calculations)
Calculation results
HCP site
Table 1 shows the calculation results at HCP site. δx means movement of H atom in x direction. δy and δz have similar meaning. δx=δy=δz=0 means the result of geometry optimization. ‘1’ means one movement which is 0.04Å.
Table 1.
| δx | δy | δz | Energy (eV) |
| 0 | 0 | 0 | -2.01835196E+004 |
| 1 | 0 | 0 | -2.01834837E+004 |
| 1 | 1 | 0 | -2.01834811E+004 |
| 1 | 0 | 1 | -2.01835046E+004 |
| 0 | 1 | 0 | -2.01834837E+004 |
| 1 | 1 | 0 | -2.01834811E+004 |
| 0 | 1 | 1 | -2.01835050E+004 |
| 0 | 0 | 1 | -2.01835070E+004 |
| 1 | 0 | 1 | -2.01835046E+004 |
| 0 | 1 | 1 | -2.01835050E+004 |
| -1 | 0 | 0 | -2.01834834E+004 |
| -1 | -1 | 0 | -2.01834815E+004 |
| -1 | 0 | -1 | -2.01834479E+004 |
| 0 | -1 | 0 | -2.01834837E+004 |
| -1 | -1 | 0 | -2.01834815E+004 |
| 0 | -1 | -1 | -2.01834479E+004 |
| 0 | 0 | -1 | -2.01834499E+004 |
| -1 | 0 | -1 | -2.01834479E+004 |
| 0 | -1 | -1 | -2.01834479E+004 |
Mass-weighted Hessian matrix calculated based on data in Table 1:
[ 4.31362677e+29 2.29142726e+29 2.59356062e+29]
[ 2.29142726e+29 4.29567826e+29 2.58159494e+29]
[ 2.59356062e+29 2.58159494e+29 4.92387633e+29]
Eigenvalues of the matrix are 9.51368753 e+29, 2.01335272 e+29, 2.00614111 e+29.
Vibration frequencies of H atom at HCP site (s^-1) are 1.5524*10^14, 0.7141*10^14, 0.7129*10^14.
Zero point energy for H at HCP site is 0.6161 eV.
FCC site
Table 2 shows the calculation results at FCC site.
Table 2.
| δx | δy | δz | Energy (eV) |
| 0 | 0 | 0 | -2.01835358E+004 |
| 1 | 0 | 0 | -2.01834780E+004 |
| 1 | 1 | 0 | -2.01834759E+004 |
| 1 | 0 | 1 | -2.01835094E+004 |
| 0 | 1 | 0 | -2.01834777E+004 |
| 1 | 1 | 0 | -2.01834759E+004 |
| 0 | 1 | 1 | -2.01835091E+004 |
| 0 | 0 | 1 | -2.01835112E+004 |
| 1 | 0 | 1 | -2.01835094E+004 |
| 0 | 1 | 1 | -2.01835091E+004 |
| -1 | 0 | 0 | -2.01834783E+004 |
| -1 | -1 | 0 | -2.01834765E+004 |
| -1 | 0 | -1 | -2.01834259E+004 |
| 0 | -1 | 0 | -2.01834784E+004 |
| -1 | -1 | 0 | -2.01834765E+004 |
| 0 | -1 | -1 | -2.01834261E+004 |
| 0 | 0 | -1 | -2.01834280E+004 |
| -1 | 0 | -1 | -2.01834259E+004 |
| 0 | -1 | -1 | -2.01834261E+004 |
Mass-weighted Hessian matrix calculated based on data in Table 2:
[ 6.89821314e+29 3.56577192e+29 4.07730464e+29]
[ 3.56577192e+29 6.91017881e+29 4.08029606e+29]
[ 4.07730464e+29 4.08029606e+29 7.92127857e+29]
Eigenvalues of the matrix are 1.51030097 e+30, 3.33850851 e+29, 3.28815230 e+29.
Vibration frequencies of H atom at FCC site (s^-1) are 1.9559*10^14, 0.9196*10^14, 0.9126*10^14
Zero point energy for H at FCC site is 0.7833 eV.
Conclusion:
Table 3 shows the energies of H at two three-hold sites on Cu(111) without and with zero point energy.
Table 3
| Energy (eV) | HCP | FCC | E(HCP) – E(FCC) |
| Without ZPE | -20183.5196 | -20183.5358 | 0.0162 |
| With ZPE | -20182.9034 | -20182.7525 | -0.1509 |
When zero point energy is not included, the H atom on FCC site has lower energy. But when zero point energy is included, the H atom on HCP site has lower energy. Zero point energy is important for light atoms, like H and should be considered for systems involving light atoms.
Reference:
- J. P. Perdew, K. Burke, and M. Ernzerhof Phys. Rev. Lett. 77, 3865 (1996)
- First principles methods using CASTEP” Zeitschrift fuer Kristallographie 220(5-6) pp. 567-570 (2005) S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson, M. C. Payne
- R. Ryberg, Carbon-Monoxide Adsorbed on Cu(100) Studied by Infrared-Spectroscopy, Surf. Sci. 114 (1982), 627 641.
