Introduction
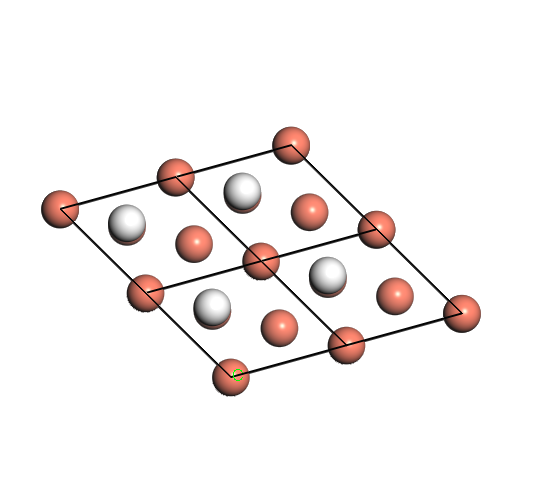
This post is aimed at exploring the convergence of k points and cutoff energy. The model chosen here is one hydrogen atom on copper(111) surface HCP site. The model of copper surface in one periodic volume is 1*1 unit cell in x-y plane and 4 layers in z direction. Vacuum is in z direction and its thickness is 10 Å.

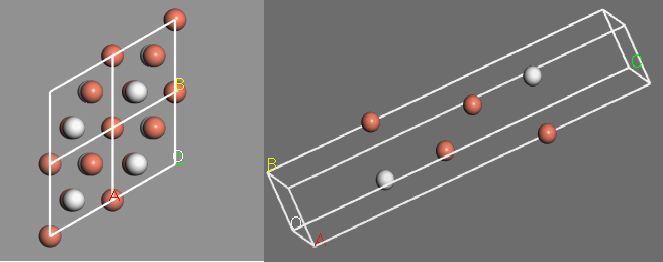
Fig. 1 A top view of the model
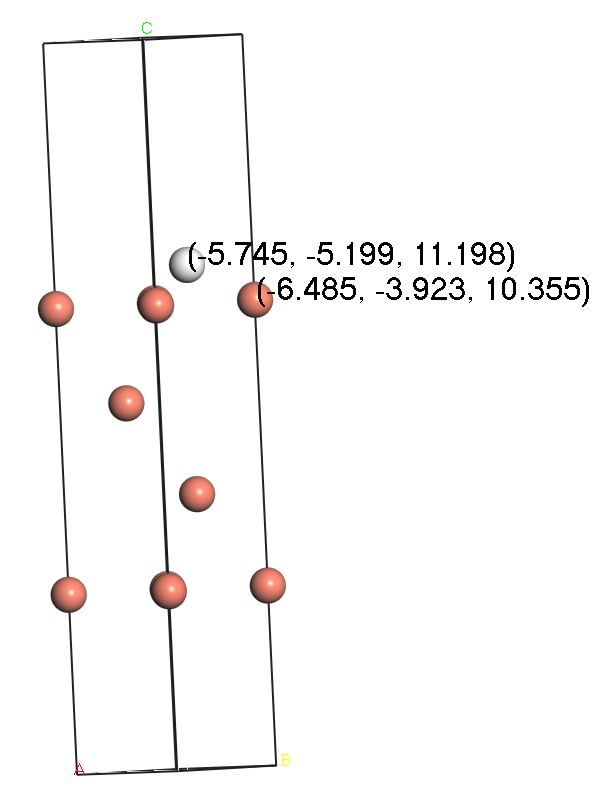
 Fig. 2 A side view of the model
Fig. 2 A side view of the model
CASTEP tool package is adopted for all calculations in this post [1].
For the purpose of simplifying the model and reducing computational cost, during geometry optimization, all copper atoms and x, y coordinates of H are fixed and H is just allowed to move in z direction.
The initial calculation setting for geometry optimization is:
Functional: GGA PBE [2]
Pseudopotentials: OTFG ultrasoft
Relativistic treatment: Koelling-Hamon
SCF tolerance: 5*10^-7 eV/atom
Max SCF cycles: 100
Dipole correction: not applied
Cutoff energy: 600 eV
K points: 11*11*2
After geometry optimization, a series of finite difference calculations are conducted by only changing cutoff energy and k points. The movement step adopted here is 0.05Å in z direction, up and down respective to equilibrium position. The energy difference of three positions is adopted as convergence indicator, named as ΔE.
Since the only freedom here is H movement in z direction, we define the indicator ΔE to be:
\begin{equation} \Delta E = E(+ \delta z) + E (- \delta z) – 2 E_0 \end{equation}
E0 is the energy for the system at equilibrium. E(+δz) means the energy of the system after H moving away from the copper surface in z direction with one step (0.05Å). E(-δz) has the opposite meaning.
The indicator is the numerator of mass-weighted Hessian Matrix and since the denominator is constant for this model the indicator shows the convergence of vibration frequency of H actually.
Results
Cutoff energy
K mesh is fixed at 11*11*2, and just cutoff energy is changed.
Calculation results are shown in Table 1. and Fig. 1. All energies are in the unite of eV.
| E_cut | 1/E_cut | E0 | E(+δz) | E(-δz) | ΔE |
| 300 | 0.003333333 | -6734.207987 | -6734.201406 | -6734.203442 | 0.011127427 |
| 350 | 0.002857143 | -6737.770064 | -6737.765333 | -6737.763871 | 0.010923297 |
| 400 | 0.0025 | -6738.585562 | -6738.581103 | -6738.578878 | 0.011143039 |
| 450 | 0.002222222 | -6738.741297 | -6738.736179 | -6738.735176 | 0.011238688 |
| 500 | 0.002 | -6738.76858 | -6738.763114 | -6738.7628 | 0.011244911 |
| 550 | 0.001818182 | -6738.777772 | -6738.772113 | -6738.772165 | 0.011265619 |
| 600 | 0.001666667 | -6738.78372 | -6738.777944 | -6738.778252 | 0.011242922 |
| 700 | 0.001428571 | -6738.789645 | -6738.783725 | -6738.784317 | 0.011249237 |
Table 1
 Fig. 3 the plot of ΔE vs reciprocal of cutoff energy
Fig. 3 the plot of ΔE vs reciprocal of cutoff energy
So A numerical guess rather than a strictly theoretical conclusion based on these data points is when we have an infinitely large cutoff energy for a fixed geometry with some specific setting, we should converge to a certain number for our indicator ΔE. When the cutoff energy goes smaller, we will have more numerical noise and the numbers will potentially deviate away from the ‘true value’ which is given by the infinitely large cutoff energy. The guess is visualized in Fig. 2.
 Fig. 4 a schematic plot showing the extreme situation of cutoff energy’s effect on ΔE
Fig. 4 a schematic plot showing the extreme situation of cutoff energy’s effect on ΔE
A part of Fig. 2 that is zoomed up to show details is shown in Fig. 3.
 Fig.5 a detailed part of Fig. 2
Fig.5 a detailed part of Fig. 2
As we can see from Fig. 3, a smaller cutoff energy will potentially give a larger deviation from the ‘true value’. Or smaller cutoff energies will give a larger range of deviation from the ‘true value’.
The exact pattern may be dependent on the indicator of convergence we choose. The practical implementation of cutoff energy in CASTEP codes will affect the pattern as well. But the tendency could be general for cutoff energy’s convergence path.
And for this specific model, the difference of ΔE from 600eV to 700eV is around 0.000006 eV. If our tolerance for ΔE is at the magnitude of 0.00001 eV, choosing 600eV as our cutoff energy under the condition of using 11*11*2 for k points is acceptable. And the converged ΔE could be 0.01124 eV.
K points
Cutoff energy is fixed at 600 eV, and just k points is changed.
First, a test for k points in z direction is conducted. The results are shown in Table 2. All energies are in the unit of eV. The difference of energies calculated from k*k*2 and k*k*1 is at the magnitude of 10^-5 eV. k means the number of k points in x or y direction. If we raise our tolerance for energy difference up to 0.0001, it could be acceptable to use ‘1’ for z direction and just change the number of k points in x and y direction for following calculations in order to reduce the computational cost.
| k mesh | E0 | k mesh | E0 | energy difference between k*k*2 and k*k*1 |
| 7x7x1 | -6738.709562 | 7x7x2 | -6738.709586 | -2.4000E-05 |
| 8x8x1 | -6738.731698 | 8x8x2 | -6738.73171 | -1.2075E-05 |
| 9x9x1 | -6738.75047 | 9x9x2 | -6738.750535 | -6.4864E-05 |
| 10x10x1 | -6738.769096 | 10x10x2 | -6738.769144 | -4.8546E-05 |
| 11x11x1 | -6738.783685 | 11x11x2 | -6738.78372 | -3.5046E-05 |
| 12x12x1 | -6738.775101 | 12x12x2 | -6738.775133 | -3.2509E-05 |
| 13x13x1 | -6738.800963 | 13x13x2 | -6738.800992 | -2.8743E-05 |
| 14x14x1 | -6738.774423 | 14x14x2 | -6738.774454 | -3.1116E-05 |
| 15x15x1 | -6738.795033 | 15x15x2 | -6738.795057 | -2.4665E-05 |
Table 2a
In order to make the convergence test for k points in z direction more convincing, we calculate 7*7*3 as well.
| k mesh | 7*7*1 | 7*7*2 | 7*7*3 |
| E0 | -6738.709562 | -6738.709586 | -6738.709615 |
| Energy difference from 7*7*1 | 0 | 2.4E-05 | 5.3E-05 |
Table 2b
As we can see, the energy difference between 7*7*3 and 7*7*1 is still at 10^-5 magnitude.
Results for a series of k*k*1 calculations are shown in Table 3 and Fig. 4. Energies are in the unit of eV.
| k-mesh | k | 1/k | E0 | E(+δz) | E(-δz) | ΔE |
| 3*3*1 | 3 | 0.333333333 | -6739.47838 | -6739.489468 | -6739.45571 | 0.011580839 |
| 5*5*1 | 5 | 0.2 | -6738.742468 | -6738.746554 | -6738.727462 | 0.010920861 |
| 7x7x1 | 7 | 0.142857143 | -6738.709562 | -6738.706263 | -6738.702423 | 0.010438667 |
| 9x9x1 | 9 | 0.111111111 | -6738.75047 | -6738.743514 | -6738.746635 | 0.010791582 |
| 11x11x1 | 11 | 0.090909091 | -6738.783685 | -6738.777909 | -6738.778218 | 0.01124229 |
| 13x13x1 | 13 | 0.076923077 | -6738.800963 | -6738.796022 | -6738.79491 | 0.010994729 |
| 15x15x1 | 15 | 0.066666667 | -6738.795033 | -6738.791071 | -6738.788042 | 0.010951909 |
| 16x16x1 | 16 | 0.0625 | -6738.770667 | -6738.766317 | -6738.763964 | 0.011054494 |
Table 3

Fig. 6 the plot of ΔE vs 1/k
Based on data points in Fig. 4, we can see the convergence of k points has a similar pattern with cutoff energy. Difference of ΔE from 15*15*1 to 16*16*1 is still at the magnitude of 10^-3 eV, which is not desirable. We can do the fitting to get the converged number of ΔE, the ‘true value’, given by infinitely large k points. The candidate fitting function could be a*exp(bx)*sin(c*x) and a,b,c are parameters needing to be fitted. Since the number of data points here is small the fitting would be loose. Instead of calculating more data points, another cheap way of approximating the ‘true value’ is doing average for last five ΔE numbers (9*9*1, 11*11*1, 13*13*1, 15*15*1, 16*16*1). The average is 0.0110070008 eV and if we truncate it to 10^-4 eV, we have 0.0110 eV, which is different from above-mentioned number, ‘0.01124 eV’, calculated from the setting of 11*11*2 as k points and 600 eV as cutoff energy. The inconsistency of using k*k*2 and k*k*1 in different tests could result in some difference. But as mentioned before, the difference should be minor. The main reason might be the average is adopted here. The number of converged ΔE could be 0.011 eV with the cost of losing some precision.
Conclusion
Based on the ΔE = 0.011 eV, vibration frequency of H on Cu(111) surface HCP site in z direction is 3.266*10^13 Hz.
Regardless of particularity of codes and models, we can say that the energy difference ΔE defined above calculated from DFT methods by using a specific k points and cutoff energy can only be trusted in a range around that number given by the infinitely large k points and cutoff energy. The larger the k points and cutoff energy is, the narrower the range is.
Acknowledge
The optimized geometry for all calculations in this post is provided by Mark Hagemann. Mark Hagemann and Brett Green also did calculations for E0 in Table 1; calculations in Table 2; calculations for E0 in Table 3 from 7*7*1 to 16*16*1.
The energy numbers for 7*7*1, 7*7*2 shared by Mark were ‘final energy(E)’ but other energy numbers are ‘final free energy(E-TS)’. So I changed Mark’s data for E0 at 7*7*1, -6738.689838(E), to -6738.709562(E-TS) for consistency.
Thank Mark and Brett for sharing their data.
Reference
- First principles methods using CASTEP” Zeitschrift fuer Kristallographie 220(5-6) pp. 567-570 (2005) S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson, M. C. Payne
- J. P. Perdew, K. Burke, and M. Ernzerhof Phys. Rev. Lett. 77, 3865 (1996)

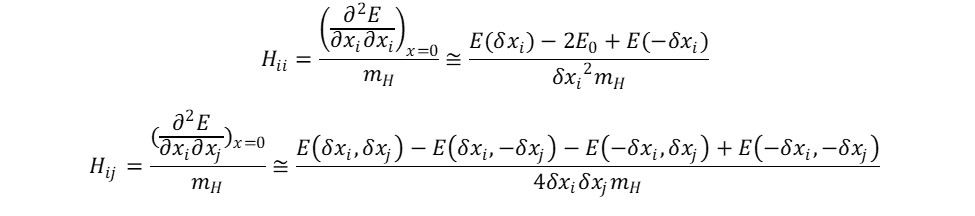











 = \frac{E(\delta x_i, \delta x_j) - 2E(x_0)+ E(-\delta x_i, -\delta x_j)}{\delta x_i \delta x_j}")


 , where found the normal mode frequencies,
, where found the normal mode frequencies,  , were calculated using the following relation.
, were calculated using the following relation.