
Abstract
In this paper, we report a systematic method of calculating phase diagram for metal hydride compound using DFT. While our calculation is limited to perfect crystal phase, it is not hard to generalize this method to more complicated systems that could even contain defects, surface phases, etc.
Structure of Li, LiH and Hydrogen Molecule
The lithium is body-centered cubic (bcc) with lattice constant 3.509Å[4] and its hydride LiH is NaCl type face-centered cubic (fcc) with lattice constant 4.007Å [1] . DFT calculations were performed to get the optimized lattice constant for LiH and the total internal energy of the lattice for both crystals, shown in figure 1.
DFT calculation is also performed to obtain the total internal energy of hydrogen molecule. This is done by fix the hydrogen molecule at a very big cubic lattice(10Å by 10Å by 10Å) and perform CASTEP calculation (figure 2). Details of calculation can be found in appendix.

Figure 1 Structure of Lithium and Lithium hydride. Li is bcc lattice while LiH is NaCl structure.

Figure 2 Hydrogen molecules. The energy is calculated by performing DFT in simple cubic lattice. The inter-molecule distance is far more greater than the size of the molecule, and the bond distance between hydrogen atoms is 0.7414Å[1].
Grand Potential for LiH and Li
In general, the grand potential of metal hydride obains the following form[2]:
\begin{equation}\Omega=E_i-T S_i-\mu_{O,i}N_{O,i}-\Omega^M\end{equation}
The first term is the energy of the metal or metal-hydride. The second term represents the effect of entropy of crystals. The lowest order approximation is to take the entropy of the crystals zero. The third term is the chemical potential of hydrogen, which depends on the number of hydrogen atoms in the crystal. The last term is a constant for two phases in our report, which is the same since both are lithium atoms. So it could be taken as zero. To be explicit, the grand potential for LiH and Li are
\begin{equation}\Omega_{LiH}=E_{LiH}-\mu_{H,i}N_{H, LiH}\end{equation}
\begin{equation}\Omega_{Li}=E_{Li}\end{equation}
In this report, we compute the grand potential per metal atom. Hence 
\begin{equation}E_{Li}=-202.44eV\end{equation}
\begin{equation}E_{LiH}=-219.13eV\end{equation}
At equilibrium, the grand potential for LiH and Li are equal. By setting two equations equal to each other, one obtains the following chemical potential relationship:
\begin{equation}\mu_{H}=\frac{1}{2}\mu_{H_2}=\frac{E_{LiH}-E_{Li}}{N_{H,LiH}}=-16.69eV\end{equation}
The chemical potential of hydrogen molecule could be evaluated by standard thermodynamic method:
\begin{equation}\mu_{H_2}=E_{H_2}^{total}+\tilde{\mu}_{H_2}(T, p^o)+k_B T \ln{(p/p^o)}\end{equation}
Where 
")
\begin{equation}\tilde{\mu}_{H_2}(T, p^o)=[H^o(T)-H^o(T_r)]-TS(T)-[H^o(0)-H^o(T_r)]\end{equation}
The standard value could be found in the NIST-JANAF Thermochemical Tables[3].
Phase Diagram
The phase diagram could be obtained by solving for 
\begin{equation}\ln{p/p^o}=\frac{1}{k_B T} (\mu_{H_2}-E_{H_2}^{total}-\tilde{\mu}_{H_2}(T, p^o))\end{equation}
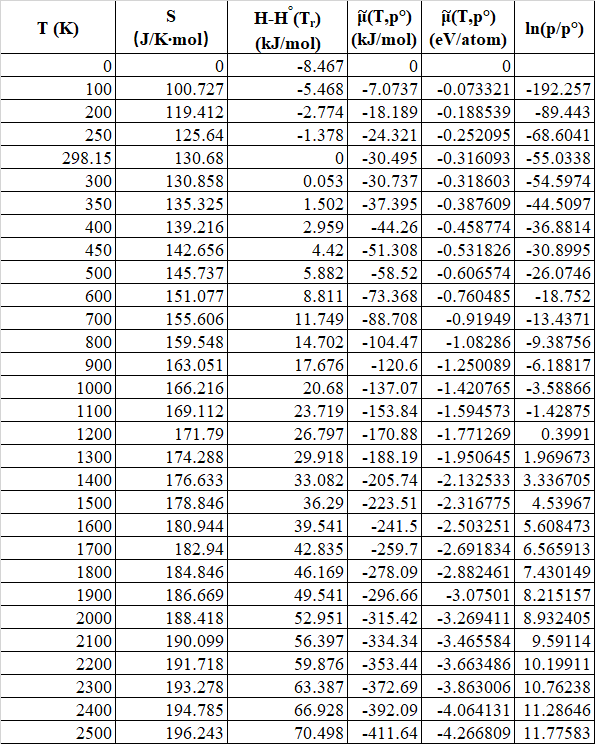
The results are shown in Table 1. It is noticeable that at low temperature and high pressure, the system tends to stay at LiH while at high temperature with low pressure, the system is more stable in Li phase. At pressure close to atmospheric pressure, the transition temperature is around 1200K, this result agrees with the experimental experience that LiH can be synthesize by Li and hydrogen at several hundreds of degrees.

Figure 3 Phase diagram of LiH/Li.
Appendix
DFT Calculation Details of Li
xc_functional : GGA PBE
spin_polarized : false
cut_off_energy : 600.0eV
calculate_stress : false
Atomic calculation performed for Li: 1s2 2s1
Pseudo atomic calculation performed for Li 1s2 2s1
number of bands : 6
k-Points For BZ Sampling: 10*10*10
DFT Calculation Details of LiH
xc_functional : GGA PBE
page_wvfns : 0
cut_off_energy : 600.0eV
Atomic calculation performed for H: 1s1
Atomic calculation performed for Li: 1s2 2s1
Pseudo atomic calculation performed for H: 1s1
Pseudo atomic calculation performed for Li: 1s2 2s1
k-Points For BZ Sampling: 6*6*6
DFT Calculation Details of Hydrogen molecule
Key parameters:
xc_functional : GGA PBE
spin_polarized : false
cut_off_energy : 450.0eV
Atomic calculation performed for H: 1s1
k-Points For BZ Sampling: 1*1*1
Lattice constants: 10.0A*10.0A*10.0A
Reference
[1] https://en.wikipedia.org/wiki/Hydrogen
[2] Sholl, David, and Janice A. Steckel. Density functional theory: a practical introduction. John Wiley & Sons, 2011.
[3] Stull, Daniel Richard, and Harold Prophet. JANAF thermochemical tables. No. NSRDS-NBS-37. National Standard Reference Data System, 1971.
[4] Dassault Systèmes BIOVIA, BIO, BIOVIA Materials Studio 2018 (18.1.0.2017), San Diego: Dassault Systèmes, 2017.
