1. Project Description
In this project, we aim to find the most stable configuration of the defective hematite (\(Fe_2\)\(O_3\)) by comparing the energy of each configuration. We modeled a 3*3*1 hematite supercell first, took one of the iron atoms out of the structure, and added three hydrogens with stoichiometry. The iron occupancy of the structure was therefore 0.991. After modeling the initial configuration, Density Functional Theory (DFT), with the Vienna Ab-initio Simulation Package (VASP) code[1], was used to optimize lattice parameter and calculate the energy. The exchange-correlation was treated in the generalized gradient approximation (GGA), as parameterized by Perdew, Burke, and Ernzerhof (PBE) [2]. GGA+U was applied to correct self-interaction of the d-electrons in Fe atoms. The following valence electron configurations were employed in the potentials: 3d6 3d7 4s1 for Fe, 2s2 2p4 for O, and 1s1 for H. Psedupotentials were used in place of other electrons, with core radii of 2.200Å for Fe, 1.100Å for O, and 0.800Å for H. The Brillouin zone integration was performed using a 1*1*1 Monkhorst-Pack k-point grid, and plane-wave kinetic energy cutoff was 800eV [3]. The SCF tolerance is 1.0e-4 eV/atom. The spin states of iron atoms in the adjacent layers were treated with opposite spin states.
2. Crystal Structure and Initial Defective Configurations
The unit cell of \(Fe_2\)\(O_3\) used Blake et al. (1970) bulk hematite structure that a=b=5.038Å, c=13.772Å, α=β=90 degrees, γ=120 degrees, and the space group is R-3c [4]. The equivalent fractional coordination in bulk hematite for iron is (0, 0, 0.35), for oxygen is (0.31, 0, 0.25). Iron occupies two-thirds of slightly distorted octahedron sites, and oxygen locates at the tetrahedron site. The Fe-O3-Fe unit repeated along the z-axis and stacked oxygens form a hexagonal close-packed (HCP) structure (Figure 1).


Figure 1. Crystal structure of hematite unit cell (\(Fe_2\)\(O_3\), Left) from Blake et al. (1970) and hematite 3*3*1 supercell (\(Fe_{108}\)\(O_{162}\), Right) after geometry optimization. View from the [110] direction. Red is oxygen; blue is iron.


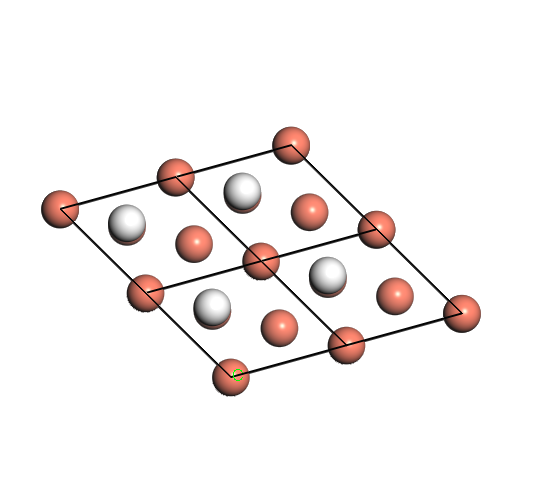
Figure 2. FeO6 octahedron (left), and O6H3 structure (right) by removing one iron and adding three hydrogens
We began with modeling one iron defect (iron occupancy 0.991) in the hematite supercell (\(Fe_{108}\)\(O_{162}\)). To compensate one defective iron (Fe3+) and reach a stoichiometry in the structure, three hydrogens were modeled and bonded with three oxygens individually (Figure 2). Because the octahedron site surrounded by six oxygens is slightly distorted in hematite structure (Figure 3), the hydrogens attached to different oxygens will generate different energy. Our goal is to find the preferred oxygen position for the hydrogen to bond with by comparing the energies of 8 different configurations.
 .
.
Figure 3. The geometry of the distorted octahedron site in hematite structure. Blue and pink atoms are iron but spin up and down respectively. Red atom is oxygen.
3. Result and Discussion
After geometry optimization and energy convergence, the energy of the 3*3*1 hematite supercell is -2002.174eV. The lattice parameters of the supercell showed that a=b=15.231Å, c=13.855Å, α=β=90 degrees, and γ=120 degrees (Figure 1). Compared to the experimental hematite structure, the computational error is 0.772%.

Figure 4. Energy convergence of configuration O 1-2-3.
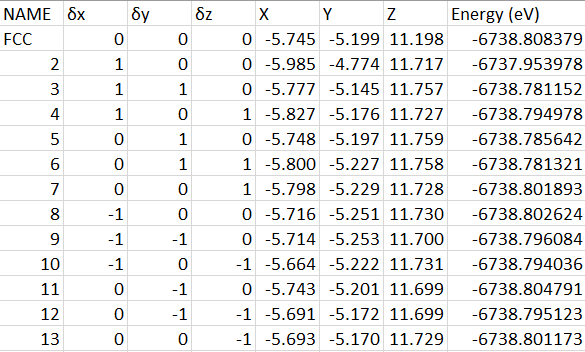
The energies of eight configurations are summarized in table 1, and one of the energy convergence plots is shown in Figure 4. The lowest energy of different configurations is -2156.654eV. The most stable structure is therefore formed where hydrogens bonded with O3, O4, and O5 (Figure 5). Accordingly, the lattice parameter of this configuration is a=15.090Å, b=15.088Å, c=13.806Å, α=β=90 degrees, and γ=120 degrees. Compared to the experimental lattice parameter of hematite, the error of the computational lattice parameter is within 0.2%.

Table 1. The energy of each configuration showed that hydrogens preferred to bond with O3, O4, and O5.


Figure 5. The most stable structure for hydrogens to fit in the octahedron before (left) and after (right) geometry optimization.
The energy differences among various configurations can be caused not only by the relative atom positions but also the hydrogen bonds. The predicted OH bond length in our DFT calculation ranges from 0.93-1.0Å, which agrees with Pinney et al. (2009) [3]. For our most stable configuration O3-4-5, the OH bond length is 0.982Å (O3H), 0.991Å (O4H), and 0.992Å (O5H). Two weak hydrogen bonds are O3H—-O2 and O3H—-O1, with bond length 2.2000Å and 2.366Å respectively. One stronger hydrogen bond formed between O5H and O1, with bond length 1.824Å.
Reference
[1] Kresse and J. Furthmuller, Phys. Rev. B, vol. 54, 1996, pp. 11169-11186.
[2] P. Perdew, B. Kieron and E. Matthias, “Generalized gradient approximation made simple,” Physical review letters, vol. 77, no. 18, p. 3865, 1996.
[3] Pinney, N., Kubicki, J. D., Middlemiss, D. S., Grey, C. P., & Morgan, D. (2009). Density functional theory study of ferrihydrite and related Fe-oxyhydroxides. Chemistry of Materials, 21(24), 5727-5742.
[4] L. Blake, R. E. Hessevick, T. Zoltai and L. W. Finger, “Refinement of hematite structure,” American Mineralogist, vol. 51, p. 123, 1966.