
1. Project Description
This project aims to calculate the vibrational frequencies and zero point energy (ZPE) of one hydrogen absorbed on face centered cubic (fcc) site and hexagonal close packed (hcp) site of Cu (111) surface. Energy calculation was performed using density functional theory (DFT) with CASTEP[1]. Exchange -correlation was treated in the generalized gradient approximation (GGA), as parameterized by Perdew, Burke, and Ernzerhof (PBE) [2]. The following valence electron configurations were employed in the potentials: 1s1 for H, and 3d10 4s1 for Cu. Psedupotentials were used in place of other electrons, with core radii of 1.996Å for Cu, and 0.599Å for H. The Brillouin zone integration was performed using a 9*9*1 Monkhorst-Pack k-point grid, and energy cutoff is 450eV. The SCF tolerance is 2.0e-6 eV/atom, and self-consistent dipole correction along the Z-axis is applied to the asymmetric slab.
2. Geometry optimization and energy calculation of two initial configurations
We will calculate the energies of hydrogen atoms adsorbed on two different threefold sites, the face centered cubic (fcc) site and hexagonal close packed (hcp) site, on Cu (111) surface. For each initial fcc and hcp configuration, we built four layers of Cu atom in the 10 Å slab and place one H atom near the surface (Figure 1&2). Then we would optimize geometry with bottom three layers of Cu fixed, and both first layers of Cu and hydrogen moved freely. After geometry optimization of the structure, the energy is calculated with the most stable relative Cu and hydrogen positions.
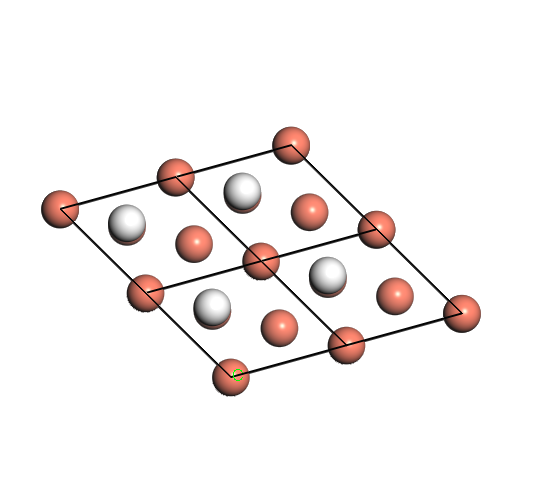

Figure 1. Top-down view of hydrogen atoms absorbed on FCC (Left) and HCP (Right) site of Cu (111) surface
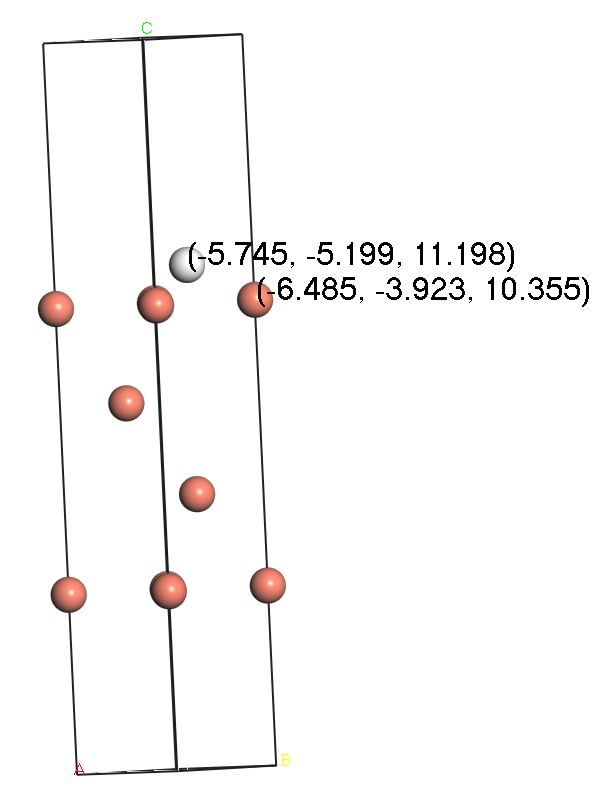

Figure 2. Side view of hydrogen atoms absorbed on FCC (Left) and HCP (Right) site of Cu (111) surface after surface relaxation
3. Hessian Matrix
We aim to calculate the Hessian Matrix by deriving energies of various configurations with slight hydrogen displacement. Sholl and Steckel (2009) suggest a good finite-difference displacements range from 0.03-0.1Å [3]. In this project, we use finite-difference with 0.06 Å along x, y, and z direction, and energies will be calculated in 12 different configurations. The minimum energy for HCP and FCC configurations are -6738.805279 eV and -6738.808379 eV; and the atom position with minimum energy is marked with δx=δy=δz=0 (Table 1).

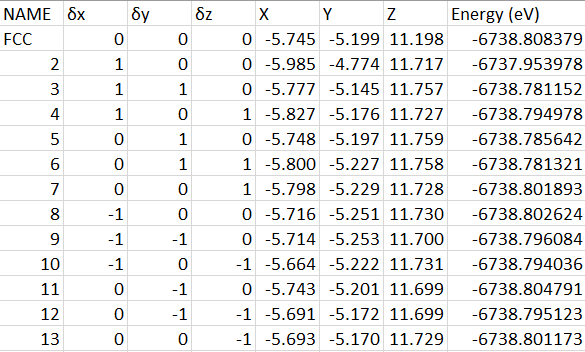
Table 1. Energies of displacements in HCP (Upper) and FCC (Lower) configurations, with a finite-difference of 0.06 Å
The Hassian matrix is calculated by second derivative of energies with displacement [3], for diagonal element, the Hessian matrix is calculated by:
\begin{equation} H_{ii}=(\frac{\partial^2 E}{\partial x_{i}^2}) \simeq \frac{E(x+\delta x_{i})+E(x-\delta x_{i})-2E_{0}(x)}{\delta x_{i}^2} \end{equation}
For off-diagonal element, the Hessian matrix is calculated by:
\begin{equation} H_{xy}=(\frac{\partial^2 E}{\partial x \partial y}) \simeq \frac{E(x+\delta x, y+\delta y)+E(x-\delta x, y-\delta y)-E(x-\delta x, y+\delta y)-E(x+\delta x, y-\delta y)}{4\delta x\delta y} \end{equation}
Thus the Hessian matrix for HCP and FCC configurations are:

4. Vibrational frequencies and zero point energy (ZPE) calculation
We can calculate the vibrational frequencies and zero point energy from the mass-weighted Hessian Matrix A, where the elements in the matrix A are [3]:
\begin{equation} A_{ij} = H_{ij} / m_i \end{equation}
The eigenvectors e and eigenvalues λ of matrix A satisfy Ae = λe. And vibrational frequencies νi are obtained from the eigenvalues λi of the matrix product A[3],
\begin{equation} \nu_i = \sqrt{\lambda}/2\pi \end{equation}
Thus the eigenvalues and vibrational frequencies of the HCP and FCC mass-weighted Hessian matrix are (Table 2):

Table 2. The calculated eigenvalue and frequencies in HCP and FCC configurations
and zero point energy (ZPE) is obtained by [3]:
\begin{equation} E_{ZPE} = \Sigma_i \frac{ h \nu_i}{2} \end{equation}
Thus the zero point energy of HCP and FCC configurations are 0.175347eV and 0.623748eV.
5. Conclusion
The energies are summarized in Table 3. The energy differences between HCP and FCC configurations without ZPE are 0.003100eV, with ZPE are -0.445301eV. That’s to say, Without including ZPE, hydrogen on the FCC site has a lower energy than HCP sites (0.003100eV), which means hydrogen in FCC site is more stable. With ZPE included, hydrogen on the FCC site has a higher energy than HCP (0.445301eV), which means HCP site is more forved. Therefore, light atoms, such as hydrogen, should consider the influence of the zero point energy.

Table 3. A summary of energy with(out) ZPC when hydrogen absorbed on HCP and FCC sites.
Reference
[1]. S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson, M. C. Payne, “First principles methods using CASTEP”, Zeitschrift fuer Kristallographie 220(5-6) pp. 567-570 (2005)
[2]. J. P. Perdew, K. Burke and M. Ernzerhof, “Generalized gradient approximation made simple”. Phys. Rev. Lett. 77, 3865 (1996).
[3]. Sholl, D. S., & Steckel, J. A. (2009). What is density functional theory? (pp. 1-33). John Wiley & Sons, Inc.
