
In this project, the absorption of H atom in the Cu (111) surface is analyzed and calculated. The H atom absorbed on Cu(111) can bind in two distinct threefold sites, the fcc sites and hcp sites shown in Figure 1. The energy difference between these two sites are calculated using Castep and the vibrational frequencies of H in each site is analyzed by calculating the elements of the Hessian matrix using finite-difference approximations. Then based on the vibrational frequencies, the zero-point energy can be obtained.

Figure 1. The absorption of H atom in the Cu (111) surface. the grey atoms are the H atoms and the red atoms are the Cu atoms. The H atoms are in the hcp sites (a) and fcc sites (b).
1. The slab model
As shown in Figure 1, an asymmetric slab model of 4 layers are used for the calculation. During the calculation, the two bottommost layers are fixed at bulk positions and all remaining atoms allowed to relax at z direction to obtain the optimized structures.
2. The optimized structures
For both two sites, the geometry optimization in the Castep is used to obtain the optimized structures, which are shown in Figure 2. We can also get the energy difference between these two structures. for hcp structure, the energy is -6738.743eV. For fcc structure, the energy is -6738.755eV. So the energy difference is 0.012eV. And since the energy of hcp structure is larger than the fcc structure, the fcc structure is more stable than the hcp structure.
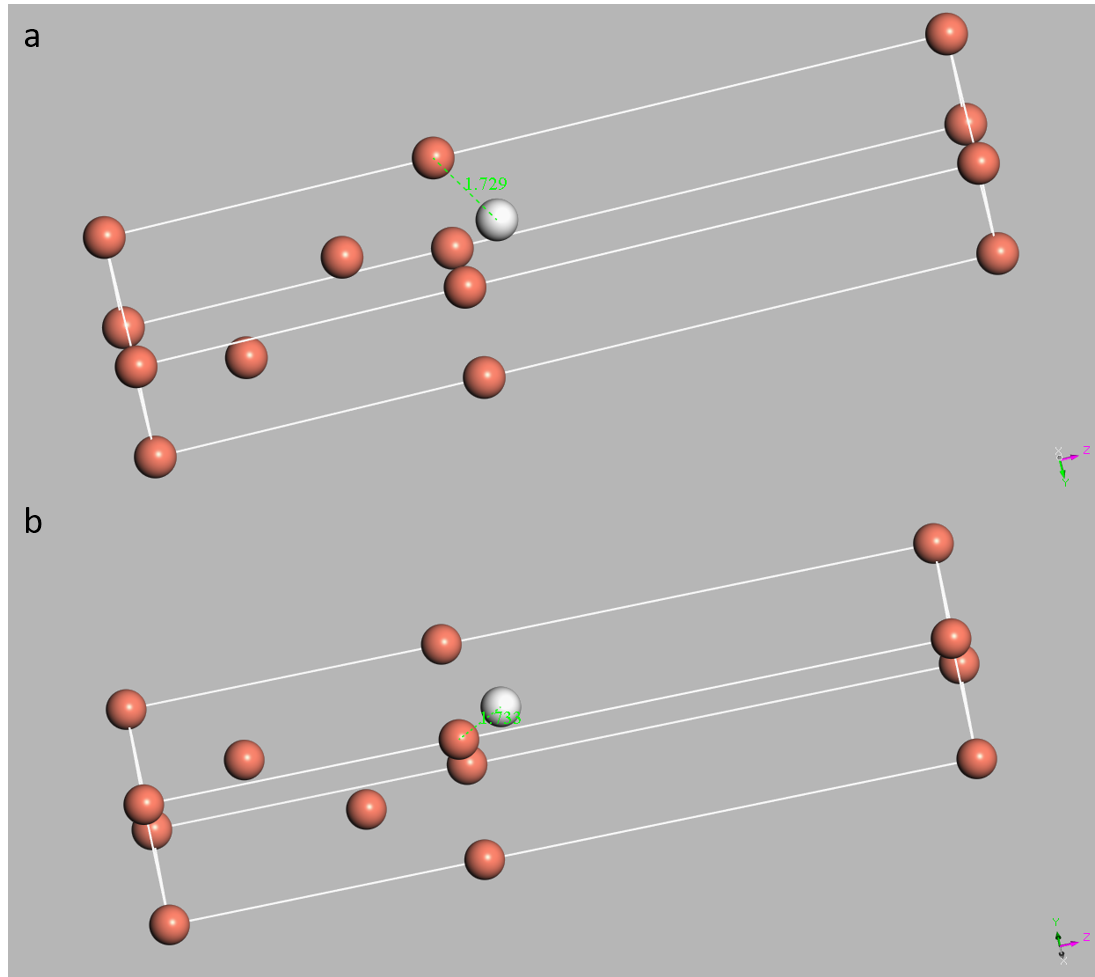
Figure 2. The optimized structures for H atoms at the hcp sites (a) and fcc sites (b). The distances are 1.729 Ångstrom in a and 1.733 Ångstrom in b.
The calculation parameters for geometry optimization are listed here:
Atomic and pseudo atomic structure for H: 1s1
Atomic structure for Cu: 1s2 2s2 2p6 3s2 3p6 3d10 4s1
pseudo atomic structure for Cu: 3d10 4s1
Functional: GGA PBE
Cut off energy: 408.2eV
k-point set: 9*9*1
Energy convergence tolerance: 1.0e-005eV/atom
Force convergence tolerance: 0.03eV/Å
Stress convergence tolerance: 0.05GPa
Displacement convergence tolerance: 0.001Å
3. The vibrational frequencies
To obtain the vibrational frequencies, we need to get the Hessian matrix first. Using finite-difference approximations, the elements in the Hessian matrix can be obtained by the equation:
Table 1 shows the calculated energy and the Hessian matrix. Then the frequencies can be obtained by the equation:![]()
λi is the eigenvalue of Hessian matrix. So we can get the frequencies shown in Table 2. The vibrational frequencies can be seen from the wave numbers of the vibrations. All the energy shown in these two table have 3 significant digits after the decimal place because we can already see the energy differences. All the Hessian matrix are shown in SI.

Table 1 The calculated energy and the Hessian matrix for hcp structure (a) and fcc structure (b).

Table 2 The eigenvalues of Hessian matrix, the vibrational frequencies and the zero-point energy for hcp structure (a) and fcc structure (b).
4. The zero-point energy correction
Since we have get the vibrational frequencies, the zero-point energy can be calculated by the equation E=∑(hv)/2.So as shown in Table 2, the total zero-point energy for hcp structure is 0.185eV and the total zero-point energy for fcc structure is 0.192eV. Then after considering the zero-point energy, the energy difference between fcc and hcp structures is 0.0046eV.
5. Conclusion and Discussion
In this project, the absorption of H atoms binding in two distinct threefold sites, the fcc sites and hcp sites on Cu (111) surface is calculated. Without considering the zero-point energy, the energy difference for these two structures is 0.012eV. With considering the zero-point energy, the energy difference for these two structures is 0.0046eV.
In this project, I assume that the H vibrations were uncoupled from the metal atoms in the surface and calculate the vibrational frequencies only moving the H atom. But this assumption is too simple and the vibrations should be more complicated in a real system. For example, we can consider the coupling between H atom and neighboring Cu atoms, which would be an interesting project for the future.
6. Reference
[1] S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson, M. C. Payne, “First principles methods using CASTEP”, Zeitschrift fuer Kristallographie 220(5-6) pp. 567-570 (2005)
