
Lev Krainov
Introduction
In the previous post[6] we found AA and AB locally stable configurations of bilayer graphene and their energies. It is well known that AB bilayer graphene is lower in energy and AA stacked bilayer transforms into it. Such a transition has to have a transition state which is the focus of this study.
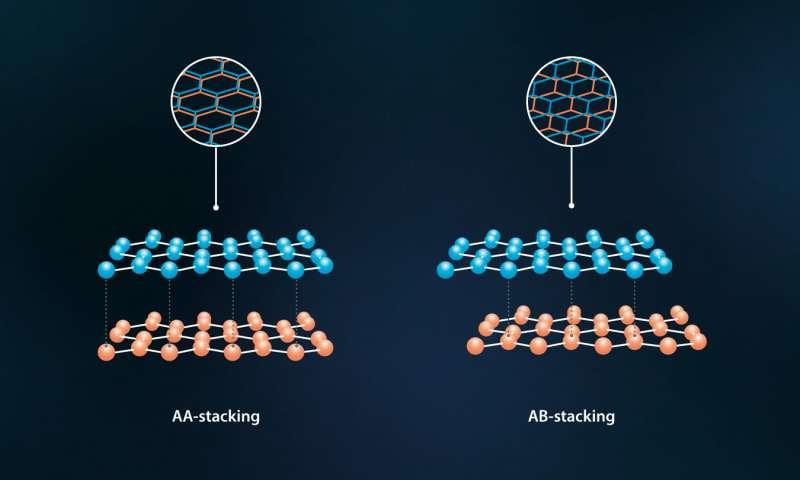
Figure 1. Bilayer graphene. AA and AB stacking. (Source [5])

Figure 2. Top(blue atoms) and bottom(orange) layers of reactant(left, AA stacking) and product(right side, AB stacking) used in calculations. Such choice avoids trajectories crossing or touching boundary of the unit cell. The unit cell is marked by a parallelogram.
Computational details
Ground state energy computations were performed using DFT plane-wave pseudopotential method implemented in CASTEP[2]. With CASTEP, we use the GGA-PBE as an exchange-correlation functional [3]. We also employ On-the-fly generated (OTFG) ultrasoft pseudopotential was used to describe the interactions of ionic core and valance electrons with a core radius of 1.4Bohr(0.74 Å) [4]. Pseudo atomic calculation is performed for 2s2 2p2 orbitals of C atoms. SCF convergence tolerance was set to 5.0E-7eV/atom. Since plane-wave method requires periodic boundary conditions we make a box of height h=17 Å and a cross section matching unit cell of graphene(a=2.46 Å).
To account for interlayer van der Waals(vdW) interaction we used Tkatchenko-Scheffler DFT-D correction[1].
Transition state search algorithm
To find transition state we employ Linear and Quadratic Synchronous Transit methods(LST/QST)[7]. The reactant and the product have to be geometrically optimized, which was done previously[6]. The parameters used for optimization were ECUT=650eV and KPOINTS=17x17x1.
The LST/QST algorithm requires one-to-one matching of product and reactant atoms. The bottom layer is trivial, but the top layer could be matched in at least two different ways. We chose matching pattern corresponding to shifting top layer as a whole, as mentioned above.
The algorithm starts with LST, followed by conjugate gradient minimization. The obtained approximation to the transition state is then used as an intermediate state for QST. The result of QST maximization is again refined by conjugate gradient minimization(CG). Then QST and CG steps are repeated until the RMS force is within 0.1eV/A. To speed up the computations we used ECUT=500eV and KPOINTS=11x11x1, providing convergence of energy up to 0.005eV.
Results
Before the transition state search both AA and AB stacked bilayers were geometrically optimized using the same ECUT and KPOINTS. The reaction energy 13meV/atom was consistent with the binding energy differences obtained previously in the studies using the same method for vdW interaction[6, 8].
The results of LST/QST search are shown on Fig 3. The search fails at LST stage because AA stacking energy is the highest on path constructed by LST.

Figure 3. Transition state search from AB(x=0) to AA(x=1).
One of the possible reasons could be that transition state is too close to AA and LST simply jumps over it. In order to check that statement we constructed our own linear path according to
 = (1-p)x_i^{AB} + px_i^{AA}")
where 

")

Figure 4. Energy along our constructed path [latex]x_i(p)[/latex] adjusted by the energy of AA configuration. Line shows a second order polynomial fit.
Conclusion
Figure 4 suggests that AA stacked bilayer graphene is not a local maximum, but could be transition state itself, since the RMS force at AA state was 2.5meV/A. One possible scenario is that AA state is a transition state between two AB states with relative shift by a primitive unit vector. Another possible explanation is that error due to our choice of system size, pseudopotential and vdW correction does not capture shallow local minimum at AA state.
Bibliography
[1] A. Tkatchenko, M. Scheffler, “Accurate molecular van der Waals interactions from ground-state electron density and free-atom reference data”, Phys. Rev. Lett. 102, 073005 (2009).
[2] S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson, M. C. Payne, “First principles methods using CASTEP”, Zeitschrift fuer Kristallographie 220(5-6) pp. 567-570 (2005)
[3] Perdew, J. P; Burke, K; Ernzerhof, M. Phys. Rev. Lett. 1996, 77, 3865-3868
[4] CASTEP GUIDE, BIOVIA, UK, 2014. URL : http://www.tcm.phy.cam.ac.uk/castep/documentation/WebHelp/content/pdfs/castep.htm
[5] https://phys.org/news/2017-01-scientists-bilayer-graphene.html
[6] https://sites.psu.edu/dftap/2019/03/29/investigation-of-interlayer-distance-for-aa-and-ab-stacked-bilayer-graphene/
[7] Halgren, T. A.; Lipscomb, W. N. Chem. Phys. Lett., 49, 225 (1977).
[8] I. V. Lebedeva, A. A. Knizhnik, A. M. Popov, Y. E. Lozovik,and B. V. Potapkin,Phys. Chem. Chem. Phys.13, 5687(2011)
