Introduction
This post will focus on applying transition state theory to hydrogen atoms adsorbed onto the surface of Cu(111). We first use geometry optimization to find the preferred position of hydrogen on the FCC and HCP sites on the Cu(111) surface. After this, transition state calculations using the complete linear synchronous transit and quadratic synchronous transit (LST/QST) method are performed in order to find the energy barriers [1].
The Slab Model and Geometry Optimization
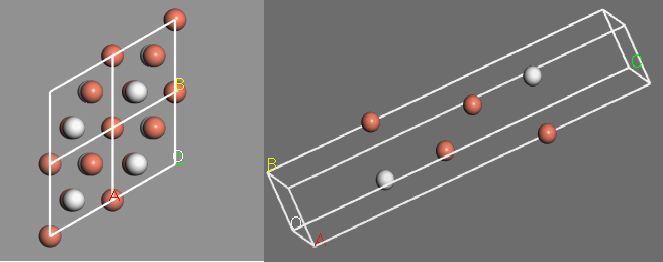
We use a slab model consisting of 4 layers of Cu and a single hydrogen atom adsorbed to the surface. The vacuum is chosen to be 10 Å thick, and the lattice vectors are (2.21 Å, -1.28 Å, 0), (0, 2.56 Å, 0), and (0, 0, 16.26 Å).

Fig 1: The slab model for Cu(111) with a hydrogen adsorbed onto the surface.
All calculations were performed with the CASTEP package in materials studio with the GGA Perdew-Burke-Ernzerhof functional [2-3]. The pseudopotential was solved with the Koelling-Harmon atomic and 1s2 2s2 2p6 3s2 3p6 for the inner shells, while 3d10 4s1 were used for the outer shells.
Geometry optimization calculations used convergence tolerances of 10^-5 eV/atom, 0.03 eV/Å for the max force, 0.05 GPa for the max stress, and a max displacement of 0.001 Å. The k-point set is increase from 6x6x1 to 10x10x1, and then from 10x10x2 to 12x12x2 to test for convergence. In changing to k-point set from 12x12x2 to 13x13x2, there is less than a 0.01 eV change in the system energy. Convergence for the cutoff energy is tested by starting at 440 eV and increasing until 570 eV. This test shows that there is less than a 0.01 eV change in the energy when going from 480 eV to 490 eV.

Fig 2: Test of the convergence for the cutoff energy. An identical trend is observed for hydrogen at the HCP site.
Transition State Calculations
The trajectory for the hydrogen transition was predicted using the Materials Studio “reaction preview” tool.

Fig 3: Side view of hydrogen moving from the HCP site to the FCC site on the surface of Cu(111) as determined by the reaction preview tool.

Fig 4: Top-down view of hydrogen moving from the HCP site to the FCC site on the surface of Cu(111) as determined by the reaction preview tool.
The transition state and energy barrier are then calculated using the LST/QST method. The RMS convergence is chosen to be 0.05 eV/Å with the maximum number of QST steps set to 5. Calculations were done to show that the energy barriers are converged well for changes of 20 eV to the energy cutoff for cutoffs past 480 eV. The reaction pathway and the energies are shown in fig 4. The transition state is found to have an energy of -6736.2477 eV.

Table 1: Variation of the energy barriers with increasing energy cutoff.

Fig 4: Reaction pathway for a hydrogen jumping from the HCP site to the FCC site. The blue star indicates the position of the transition state.
We have previously found that the hydrogen atom at the HCP site has vibrational frequencies of 36.9 THz, 32.7 THz, and 33.4 THz, while at the FCC site it has vibrational frequencies of 32.1 THz, 31.8 THz, and 31.1 THz. We can once again apply the Hessian matrix method to find the vibrational frequencies for the transition state. These frequencies give zero-point energy corrections of 0.213 eV and 0.196 eV, respectively.

Table 2: Perturbations in the energy for small changes in the hydrogen atom position at the transition state. These are used to calculate the Hessian matrix.
Doing this, we find that the transition state has real frequencies of 63.2 Thz and 32.0 THz. There is one imaginary frequency of 23.0 Thz, which corresponds to the hydrogen moving from the HCP site to the FCC site or vice-versa. The corresponding zero-point energy correction is then 0.197 eV.
Using the zero-point energy corrections, we can then find the corrections to the energy barriers. This correction is defined as
\[\Delta E_{ZP} = \bigg( E^{\dagger} + \sum_{j=1}^{N-1} \frac{h \nu_{j}^{\dagger}}{2} \bigg) – \bigg( E_{A} + \sum_{i=1}^{N} \frac{h \nu_{i}^{\dagger}}{2} \bigg) \]
Here, \(E^{\dagger} \) is the energy of the transition state and \(E_{A} \) is the energy in either the HCP site or the FCC site. The sum with j sums over the N-1 real modes at the transition state, while the sum over i sums over the normal modes in either the FCC or HCP site. These two sums simply give the zero point energy corrections that we have calculated above.
For a transition with hydrogen starting in the HCP site, the energy barrier will be corrected to 0.230 eV. For a transition from the FCC site, the correction to the energy barrier is 0.219 eV.
Conclusions
Using transition state theory, we find that hydrogen hopping from the HCP to FCC vacancy site on the surface of Cu(111) has an energy barrier of 0.247 eV, while hopping in the other direction has an energy barrier of 0.220 eV. Using the zero-point energy correction, we find that these barriers are corrected to 0.230 eV and 0.219 eV, respectively. These correspond to 4.3% and 0.5% changes from the original energy barriers.
References
[1] Govind, N., Petersen, M., Fitzgerald, G., King-Smith, D., & Andzelm, J. (2003). A generalized synchronous transit method for transition state location. Computational Materials Science,28(2), 250-258.
[2] First principle methods using CASTEP Zeitschrift fuer Kristallographie 220(5-6) pp. 567-570 (2005).
[3] Perdew, J.P., K. Burke, and M. Ernzerhof, Generalized gradient approximation made simple.Physical review letters, 1996. 77(18): p. 3865.