
When you do research, you’re not just researching your topic: you’re researching yourself, and learning how you work best. I’ve learned lab techniques this year, yes, but none is as important as these two acquired lessons: my interests are labile, and read up on lab literature before joining a lab. I want to switch labs, and I will remember these lessons when making that switch.

How did I initially decide to work in Dr. Wang’s lab? “I do research in epigenetics,” said Dr. Wang on the first day, a Monday, of my BMB 251H class last fall. “I’m interested in epigenetics,” I thought, and subsequently asked if I could visit his lab. On Tuesday, I was sitting in Dr. Wang’s office, conversing about notable undergrads with whom he had previously worked, who were now in prestigious medical or graduate programs. Starting by shadowing Jinquan in September, I embarked on her research on a protein called HDGF2. However, on the penultimate day of fall semester, I took Dr. Wang out to lunch and said that I felt that I’d been in his lab for a semester but had no real responsibilities, and that I was having trouble committing to a regular schedule. He said that I could have more responsibility next semester (as in this one, almost past) and schedule weekly lab time, just as my classes are scheduled weekly. On that optimistic note, I went on winter break.
Over break, I read some of Dr. Wang’s papers to learn background information for my research this semester. But I was also searching for summer research. After realizing that my trip to China to take BMB 252 precluded me from most summer research programs, I found an email from SHC Career Development inviting undergraduates to work in Dr. Costas Maranas’s lab. His lab uses computers to design proteins which, when incorporated into bacteria, would enable them to produce biofuels and other valuable chemicals from methane and organic waste. An incentive: Dr. Maranas was offering a stipend, while Dr. Wang was not, and subletting an apartment in State College over the summer would cost money. Thus, I emailed Dr. Maranas, read two of his papers, and met with his graduate students; they informally interviewed me and judged that I would have sufficient background knowledge and programming skills by mid-June to contribute to the project. One graduate student was applying for a grant that required that two undergrads collaborate on his project, so he added me to the list, and just like that, I was committed to the lab for the summer, the day before spring semester began.
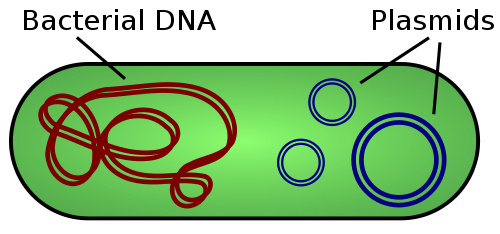
The first two weeks back in Dr. Wang’s lab were great. I had floundered the previous semester, learning (by which I mean failing, mostly) to insert a circular piece of DNA called a plasmid into E. coli, grow the E. coli to generate a lot of plasmids, and then extract the plasmids from the E. coli. The second week, though, as classes were still yawning awake, I planned how I would use every chunk of free time to do every step towards generating that DNA in E. coli. I ran to lab in the morning, set up a reaction, ran to class, to lunch, to class, and back to lab; it was stressful, like being a lab mouse in a maze. But in five days, I had two tiny tubes containing the right plasmids dissolved in water, as well as a swelling of pride: more than I had accomplished my first semester, I had done in a week. At that moment, I was quite sure I had a bright future in Dr. Wang’s lab.
That satisfaction, however, never came again. As classes accelerated, I could not manage to devote most of my non-class time in subsequent weeks to research. The plasmid extraction part of the project finished, I looked forward to the next part: putting the plasmid into bone cancer cells and measuring how it affected their gene expression via western blotting. Jinquan had to do those steps. Meanwhile, she had told me to read up on HDGF2 protein for another project, so I read some papers, but that project was on hiatus, as the mice missing one copy of the HDGF2 gene would take a month to produce mice missing both copies. (Genes are italicized; the proteins they encode are not.) I kept drifting from project to project; not committing to any one, I was not significantly learning, progressing, or feeling satisfied. The work was stressful; I never felt like I was doing well enough to please Jinquan since the week I made the plasmids. I was lonesome, working without a fellow classmate with whom I could discuss the research and have fun outside of the lab. Moreover, the specific projects of Dr. Wang and Jinquan were interesting me less and less.
What was replacing them? Microbiology, auspiciously. Before I even applied to Schreyer, I remember looking at the list of Honors courses and thinking dismissively, “Oh, I can take Honors Microbiology. Are there any classes about nutritional biochemistry?” How do you tell a good class? It changes your mind. My Microbiology 201H and Microbiology 202 lab classes have switched my interests from eukaryotic (animal, plant, fungal) to prokaryotic (bacterial, archaeal) biochemistry in only three months. Although I am still interested in human biology, I recognize now how much microbes profoundly affect human health. It startled me that our ways to identify and kill pathogenic microbes are much more tenuous than I imagined, and that resistant bacteria are on track to vitiate our last reserves of antibiotics. I thus attended a screening of The Resistance, a documentary about antibiotic resistance that would compel any biologist to switch to that field.
Microbes relate in more ways to human health. I attended a lecture here, given by University of Colorado Boulder professor Rob Knight, about how the bacteria living in our intestines profoundly affect our risk for developing obesity and other chronic diseases, but that we have only superficial knowledge of how they do this, or even what bacterial species are living there. Writing a term paper for Microbiology 201H, I learned that the microbes in our intestines even regulate our appetite. Some bacterial species not only thrive when we eat too much but also can compel us to eat too much; if they gain a stronghold, they can flourish as they make us gorge to obesity.
A little more indirectly, but no less pressingly, microbes affect human health via their potential to purify water, generate electricity, recycle waste, and produce valuable chemicals. Producing sufficient energy, water, and food are likely the three most important problems now facing humanity, even more important than cancer epigenetics (the focus of Dr. Wang’s lab), or even antibiotic resistance and obesity. If we could use unlimited energy to distribute unlimited potable water and nutritious food to everyone in the world, we would reduce malnutrition, poverty, obesity, and other chronic diseases, as well as infectious disease, which often spreads in third world nations via contaminated water, and which the aforementioned ailments exacerbate. Microbes have a role: in Microbiology 202 lab, we produced 0.3 milliwatts of electricity by packing two wires and some normal bacteria-laden dirt into a plastic jar. One Penn State professor can generate somewhat more electricity: Dr. Bruce Logan is one of the world experts on microbial fuel cells, and I am interested in visiting his lab this summer, possibly to work in his lab next fall. However, as I said at the beginning of this post, I will give his lab thought before I interview to join.
In my desire to switch labs, I have been visiting other labs and reading their background literature. Dr. Gong Chen recently made a breakthrough in repairing brain injuries; I interviewed him about and read some of his papers, but I decided that his research is not as quantitative as I would like, and that the projects his lab is moving towards—murine behavior tests—are not my passion. Dr. Miyashiro, my Microbiology 202 lab professor, is doing work with host-microbe symbiosis in squid (the human microbiome is a type of host-microbe symbiosis, too), but I visited his lab and again do not think the research is quite right. However, I am looking forward to Dr. Costas Maranas’s lab this summer. Working on a coding project this semester, I have realized that coding things on computers is one of the few activities that will keep me up until 3 AM (twice!). Add that I enjoy physics, chemistry, and biochemistry, which are integral to protein design and metabolism. Add that the mission of the lab is to design bacteria that recycle organic waste into useful chemicals, and I have more than a hunch that I will enjoy the work.
Do I know for sure? No, but I need to do research to learn if this is work I enjoy. I know my mission: make health sustainable. I do not know my approach, which is unsettling, but also exciting. As I am scoping it out, and indeed, as you too are figuring out what you want to do, I think that a good take-away message is this: always pay attention to how well the work fits you. Does it fulfill you? Aggravate you? Cut into your hours for friends, fun, or sleep? Do you have to force yourself to do it, or do other people have to pull you away from it? Don’t shoehorn yourself into something you hate, because you can find what you love. How? Search, research, and research.













#/media/File:Streptococcal_hemolysis.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
.svg){kind=link}